Regelverkene for medisinsk utstyr er spesielt viktig for KI-systemer innen helse- og omsorgstjenesten. I tillegg definerer den kommende KI-forordningen viktige prinsipper og vil stille krav til både produsenten og brukeren (f.eks. helseinstitusjonen) av KI-systemer med høy risiko [33]. Det kan være hensiktsmessig å ta hensyn til disse kravene allerede nå, spesielt for høy-risikosystemer, selv om forordningen ikke er trådt i kraft per 2024 i Norge. Personopplysningsloven og den generelle helselovgivningen utgjør også viktige rammer for bruk av KI-systemer [34].
Tiltenkt formål
Hva KI-systemet er tiltenkt for, det vil si hvilket formål systemet skal brukes til, vil være bestemmende for hvilke(t) regelverk som gjelder [35]. KI-modeller for generelle formål [36] (eng: general-purpose AI models (GPAI-models)) har ikke et tiltenkt formål, men kan inngå i et utstyr som har et medisinsk formål. De faller inn under spesifikke reguleringer i EU-forordningen for KI.
KI-systemer er medisinsk utstyr etter lov om medisinsk utstyr dersom det er ment å skulle brukes på mennesker i den hensikt å, bidra til diagnostisering, forebygging, overvåking, prediksjon, prognostisering, behandling eller lindring av sykdom [37][38]. Andre KI-systemer som for eksempel logistikksystemer og systemer for vaktplanlegging er i utgangspunktet ikke medisinsk utstyr. KI-systemer som for eksempel journalsystemer, kan imidlertid ha tilleggsfunksjoner som kan medføre at de klassifiseres som medisinsk utstyr. Koordineringsgruppen for medisinsk utstyr i EU (MDCG) [39] har publisert veiledende dokumenter som kan bidra til å vurdere når programvare faller inn under definisjonen av medisinsk utstyr [40].
- Hvis det tiltenkte formålet er medisinsk, gjelder følgende:
- KI-systemet betraktes som medisinsk utstyr og virksomheten må bruke et CE-merket utstyr [41]. Utstyret må oppfylle dokumentasjonskravene i Lov om medisinsk utstyr.
- KI-systemet vil i utgangspunktet betraktes som høyrisikosystem i henhold til KI-forordningen og må oppfylle kravene som stilles til disse systemene når den trer i kraft [42].
- Hvis det tiltenkte formålet ikke er medisinsk, gjelder følgende:
- KI-systemet betraktes som høyrisikosystem i henhold til KI-forordningen dersom tiltenkt formål er listet i vedlegg III og må oppfylle kravene i denne, når den trer i kraft [43]. Det kan for eksempel være tilfelle dersom bruk av KI-systemet vil påvirke noens rett til å motta ytelser eller helsetjenester [44]. I dette tilfellet må offentlige tjenestetilbydere også utføre en risikovurdering av fundamentale menneskerettigheter (Fundamental Rights Impact Assessment (FRIA)) [45].
- Hvis KI-systemet er ment å samhandle direkte med fysiske personer, gjelder følgende, med noen unntak:
- KI-forordningen krever transparens: KI-systemet må være designet og utviklet slik at berørte personer informeres om at de samhandler med et KI-system [46].
- I tillegg må virksomheten sjekke om KI-systemet kan falle innenfor annet produktregelverk [47]. I avsnittet Alignment of product legislation på denne websiden om New legislative framework gis en oversikt over EUs produktgrupper [48].
Lov om medisinsk utstyr
Lov om medisinsk utstyr [49] gjennomfører forordningene om medisinsk utstyr (MDR og IVDR). Dersom et KI-system skal benyttes til et medisinsk formål må det anskaffes et medisinsk utstyr. Det kan finnes lignende systemer som er satt på markedet under andre regelverk, men disse oppfyller ikke kravene til sikkerhet som stilles til medisinsk utstyr.
- Medisinsk utstyr kan kun markedsføres, distribueres eller tas i bruk når utstyret oppfyller de grunnleggende kravene til sikkerhet og ytelse etter regelverket, er samsvarsvurdert og dermed påført CE-merket, jf. håndteringsforskriften § 7. Denne forskriften pålegger at virksomheten anskaffer medisinsk utstyr som er egnet for det tiltenkte bruksområdet, og at utstyret brukes i samsvar med utstyrets vedlagte anvisninger.
Risikoklasser for medisinsk utstyr
Omfanget av samsvarsvurderingen etter lov om medisinsk utstyr avhenger blant annet av hvilken risikoklasse det medisinske utstyret tilhører [50].

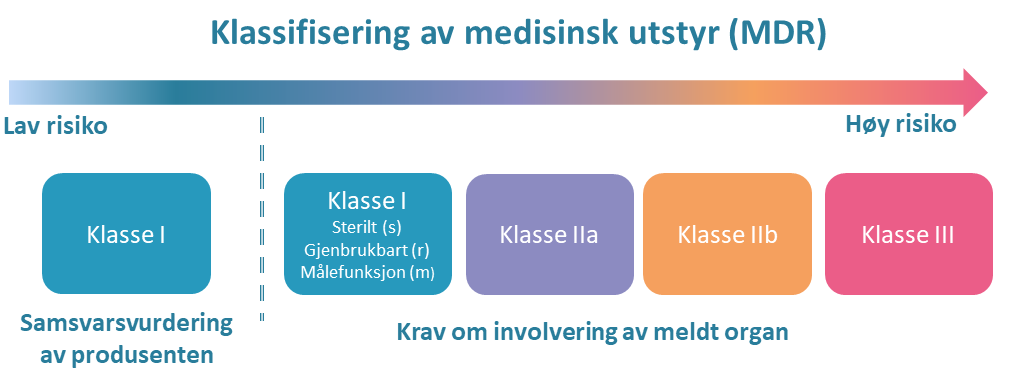{kind=link}
Medisinsk utstyr klassifiseres i fire klasser (I, IIa, IIb og III) basert på risikoen forbundet med deres tiltenkte bruk. For de fleste typer utstyr i klasse I er det tilstrekkelig at produsenter selvdeklarerer utstyret. For utstyr i klasse IIa, IIb og III, (samt utstyr i klasse Ir, Is og Im), skal det foreligge sertifikat fra meldt organ som vurderer samsvar med kravene. Det finnes en oversikt over klassifiseringsregler på DMP's nettsider [51].
Medisinsk utstyr for in vitro-diagnostikk (IVD) har egne klasser (A, B, C og D) [52]. Krav om sertifikat fra meldt organ gjelder for IVD-utstyr i klasse B, C og D.
Produsentansvar
Hvis medisinsk utstyr brukes til annet tiltenkt formål enn beskrevet i samsvarserklæringen, eller virksomheten endrer utstyret slik at det kan påvirke samsvaret med gjeldende krav, medfører det at virksomheten tar på seg produsentansvaret, altså det juridiske ansvaret for oppfyllelse av alle krav i MDR slik at KI-systemet er sikkert og trygt [53].
Et eksempel på dette fra diagnostisk radiologi [54]:
- Eksempel: Et KI-system som er utviklet for bruk på voksne kan ikke uten videre tas i bruk på barn (0-18 år). Begrunnelsen for dette er at barn ikke kan behandles som små voksne. Det kreves særskilt kompetanse for å vurdere om et KI-system er trygt å bruke på barn og om KI-systemet gir god nok ytelse på barn sammenliknet med ytelsen på voksne. Dersom tiltenkt formål for KI-systemet er diagnostisk radiologi på voksne, men man i stedet vil bruke det på barn, må KI-systemet undersøkes for aldersgruppen gjennom en klinisk utprøving. Dersom virksomheten tar i bruk et slikt KI-system på barn, tar de på seg et produsentansvar.
Helseinstitusjoner har mulighet til å produsere medisinsk utstyr for intern bruk. DMP's nettsider beskriver hvilke krav som gjelder for dette [55].
Overgangsordninger
I tiden fremover vil det fortsatt være medisinsk utstyr på markedet som følger de gamle EU-direktivene [56]. Det er innført overgangsordninger mellom de gamle og de nye regelverkene for å forhindre mangler på det europeiske markedet, se DMP's nettsider [57].
Klinisk utprøving av et KI-system som ikke er CE-merket
Dersom det ikke finnes et alternativ som er CE-merket, eller hvor et KI-system uten CE-merking vurderes som egnet, vil det i tilfelle være nødvendig å søke om tillatelse for en klinisk utprøving hos DMP og Etikkomiteene for klinisk utprøving av legemidler og medisinsk utstyr (REK KULMU) [58].
FDA-godkjenning
Medisinsk utstyr som er godkjent av de amerikanske helsemyndighetene (US Food and Drug Administration (FDA)) [59], men som ikke er samsvarsvurdert i EU og CE-merket, kan ikke lovlig brukes i Norge eller i resten av EU/EØS, og bør derfor ikke anskaffes. Det hender at produsenter har ulike versjoner eller modeller for det amerikanske og det europeiske markedet. I slike tilfeller er det viktig å sørge for at virksomheten anskaffer det CE-merkede utstyret som er ment for det europeiske markedet.
KI-forordningen
Formålet med KI-forordningen (eur-lex.europa.eu) er å fremme og øke bruk av menneskesentrerte og tillitsverdige KI-systemer, sikre et høyt nivå av beskyttelse av helse, sikkerhet og grunnleggende rettigheter, inkludert demokrati, rettssikkerhet og miljø, mot skadelige effekter som kan følge av KI-systemer og å støtte innovasjon.
KI-forordningen ble publisert 7. juli 2024 [60]. De ulike kravene i forordningen vil stegvis tre i kraft [61]. Regjeringen vil jobbe for at KI-forordningen blir innlemmet i EØS-avtalen så raskt som mulig [62].
EU-kommisjonen har bestilt europeiske harmoniserte standarder, som fungerer som et sett med tekniske spesifikasjoner som kan hjelpe med å oppfylle kravene i KI-forordningen [63].
Etter KI-forordningen er det den tiltenkte bruken av KI-systemet som bestemmer risikoklassen, ikke bare KI-systemet i seg selv. Det samme KI-systemet kan derfor ha lav risiko i én brukssituasjon, men høy risiko i en annen.
Pyramide som illustrerer forskjellige risikoklasser i KI-forordningen. Fra bunn til topp: minimal risiko (art. 95), begrenset risiko (art. 50), høyrisiko (art. 6 og 7.) og uakseptabel risiko (art.5).

Høyrisiko KI-systemer
Et høy-risiko KI-system kan etter KI-forordningen enten tilhøre kategorien regulerte produkter (som for eksempel medisinsk utstyr) eller kategorien regulerte bruksformål [64].
KI-forordningen beskriver to kategorier høyrisiko-KI-systemer
Regulerte produkter: Vedlegg I i KI-forordningen
Dersom det er krav om tredjepartssertifisering av et produkt eller sikkerhetskomponent i et produkt som allerede regulert av et av EU-regelverkene nevnt i vedlegg I, blant annet:
- Medical Device Regulation (MDR)
- In Vitro Diagnostic Device Regulation (IVDR)
- Maskindirektivet, Leketøysdirektivet m.fl.
Regulerte bruksformål: Vedlegg III i KI-forordningen
Frittstående systemer som:
- Biometrisk identifisering, følelsesgjenkjenning
- Kritisk infrastruktur
- Utdannelse og yrkestrening
- Ansettelse og arbeidsforhold
- Tilgang til grunnleggende velferdstjenester
- Rettshåndhevende myndigheter
- Migrasjon, asylsaker, grensekontroll
- Demokratiske prosesser og domstolenes virke
Kravene til KI-systemer som er eller inngår i medisinsk utstyr trer i kraft i EU 2. august 2027 [65]. Kravene til høyrisikosystemer brukt for visse formål, jf. vedlegg III, trer i kraft 2. august 2026.
KI-forordningen stiller blant annet følgende krav til høyrisikosystemer:
- Risikostyringssystem (artikkel 9)
- Data og datahåndtering (artikkel 10)
- Teknisk dokumentasjon (artikkel 11)
- Loggføring (artikkel 12)
- Transparens og informasjon til brukere av systemet (artikkel 13)
- Menneskelig overblikk (artikkel 14)
- Nøyaktighet, robusthet og cybersikkerhet (eng: Cyber Security) (artikkel 15)
Artikkel 11 beskriver krav til teknisk dokumentasjon for et hvert høyrisiko KI-system og dette er ytterligere beskrevet i forordningens vedlegg IV [66]. Dette er dokumentasjon som leverandørene vil måtte utarbeide etter hvert som deler av KI-forordningen får anvendelse i EU, men vil allerede nå kunne være til hjelp i anskaffelsesprosessen.
Begrepene transparens og menneskelig overblikk (i artikkel 13 og 14) kan være nytt å forholde seg til i anskaffelsesprosessen. At KI-systemet er transparent betyr blant annet at det er utformet på en måte som gjør det mulig å forstå hvordan KI-modellen er utviklet. Videre at det er tydelig for brukerne at de samhandler med et KI-system. Når brukeren er klar over at systemet benytter KI, kan brukeren ta hensyn til dette når hen vurderer og benytter informasjon fra systemet. Menneskelig overblikk av et KI-system kan innebære en form for menneskelig tilsyn eller mulighet for menneskelig innblanding dersom det er nødvendig. Brukeren skal ikke være helt avhengig av systemet for kritiske beslutninger. Menneskelig overblikk forutsetter blant annet riktig kompetanse og opplæring [67].
KI-systemer med begrenset risiko
Systemer med begrenset risiko er ikke underlagt de samme strenge kravene som høy-risiko KI-systemer, men det er fortsatt krav om at KI-systemene må være transparente og at KI-generert innhold skal merkes i maskinlesbart format [68]. Virksomheter som tar i bruk KI-systemer har videre plikt til å sikre tilstrekkelig kompetanse hos personellet som vil bruke eller drifte KI-systemene.
KI-modeller for generelle formål
KI-modeller som er laget for generelle formål, også kalt GPAI-modeller (eng.: General Purpose Artificial Intelligence (GPAI) [69]), kan brukes enten direkte, som ChatGPT og Gemini, eller de kan tilpasses og/eller finjusteres av andre produsenter for et spesifikt formål. Produsenter som baserer seg på GPAI-modeller, vil trenge tilstrekkelig dokumentasjon om disse modellene for selv å kunne dokumentere kvaliteten på sitt produkt.
Selv om GPAI-modellene ikke har et spesifikt formål i seg selv, blir de likevel regulert i KI-forordningen dersom de oppfyller visse vilkår i artikkel 51 [70]. Leverandører av GPAI-modeller må bl.a. sørge for at de som bruker modellene deres forstår dem godt nok til å kunne bruke dem i sine produkter og oppfylle sine forpliktelser i henhold til loven [71]. Dersom slike modeller i tillegg har systemisk risiko, gjelder strengere krav [72]. Eksempler på systemisk risiko kan være at KI-systemet har evne til å påvirke valg, kritisk infrastruktur og beredskap. Leverandører av GPAI-modeller med systemisk risiko må blant annet utføre nødvendige modellvurderinger for å identifisere, og kontinuerlig vurdere og redusere systemiske risikoer gjennom risikostyring, overvåking etter at modellen er lansert, og samarbeide med relevante aktører. Alvorlige hendelser og mulige korrigerende tiltak skal umiddelbart rapporteres til EU-kommisjonen og nasjonale kompetente myndigheter.
I påvente av harmoniserte standarder har EU-kommisjonen lansert et første utkast til retningslinjer for KI-modeller for generelle formål [73].
Andre aktuelle regelverk
KI-ansvarsdirektiv
EU-kommisjonen har fremmet et forslag til et KI-ansvarsdirektiv gjelder sivilrettslig erstatningsansvar utenfor kontrakt når skaden forårsakes av et kunstig intelligent system. Forslaget fastsetter felles regler for fremleggelse av dokumentasjon for høyrisikosystemer med kunstig intelligens for å gjøre det mulig for saksøker å underbygge et eventuelt erstatningsansvar [74].
Automatiserte avgjørelser
Personvernforordningen (GDPR) har flere bestemmelser om automatiserte individuelle avgjørelser, særlig i artikkel 22, som handler om avgjørelser som fattes uten menneskelig innblanding. Reglene i personvernforordningen vil slå inn når KI-systemer foretar slike automatiserte avgjørelser eller når de brukes med helmaskinell behandling. Innbyggere har rett til ikke å være gjenstand for en avgjørelse som utelukkende er basert på automatisert behandling, herunder profilering, som har rettsvirkning for eller på tilsvarende måte i betydelig grad påvirker vedkommende [75]. Det gjelder flere unntak, men i så fall har innbyggerne òg særskilte rettigheter, som å kreve at et menneske ser på avgjørelsen [76]. Krav om forklarbarhet og transparens angående informasjon om logikk som ligger til grunn for automatiserte avgjørelser er beskrevet i artiklene 13, 14 og 15 i personvernforordningen [77]. Bestemmelser om automatiserte avgjørelser finnes i pasientjournalloven § 11 og folketrygdloven § 21-11a, og åpner også opp for at det kan gis forskrift om at mer inngripende avgjørelser kan treffes automatisert.
Regelverk for tilgang og bruk av helseopplysninger
Helseopplysninger er etter norsk rett underlagt taushetsplikt og for all behandling av personopplysninger stilles det krav om behandlingsgrunnlag etter artikkel 6 og artikkel 9 i personvernforordningen for behandling av helseopplysninger [78].
I all behandling av person- og helseopplysninger er det krav om at det er en dataansvarlig [79]. Den dataansvarlige har ansvaret for at behandlingen av person- og helseopplysninger er i samsvar med gjeldende regelverk, blant annet å sørge for at helseopplysningene behandles lovlig og for tilfredsstillende informasjonssikkerhet og internkontroll.
Personvernforordningen gir rammer for dataansvarliges plikter og oppgaver, og legger opp til sanksjoner dersom pliktene ikke overholdes. å sørge for at helseopplysningene behandles lovlig. Andre sentrale plikter er blant annet å sørge for tilfredsstillende informasjonssikkerhet og internkontroll.
Dataansvarlig kan inngå avtale med en databehandler om behandling av personopplysninger; en databehandleravtale. Dette er aktuelt når andre skal behandle opplysningene på deres vegne. Databehandleren kan bare behandle opplysningene etter avtalen med dataansvarlig.
Se mer utfyllende om taushetsplikt og tilgang til helseopplysninger for validering i fase 5.
Strålevernregelverket
Innføring av KI-systemer kan påvirke bruk av det strålegivende utstyret eller andre forhold relevant for strålevern, for eksempel beslutningsstøtte for vurdering av berettigelse av en radiologisk undersøkelse. Mange av de aktuelle KI-systemene er tatt i bruk innen fagområder som radiologi, og stråleterapi. Strålevernregelverket stiller også strenge krav til blant annet risikovurderinger, kvalitetskontroll av systemene og kompetanse til å bruke disse [80].
[35] Definisjon av ”tiltenkt formål” i KI-forordningen art. 3 nr. 12 https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=OJ:L_202401689#d1e2090-1-1 () og MDR art. 2 nr. 12 (32017r0745.pdf (lovdata.no))
[36] Det finnes ingen offisiell norsk oversettelse. I denne rapporten lener vi oss på den danske oversettelsen av KI-forordningen, som bruker de begrepet "AI-model til almen brug".
[38] Se for øvrig EØS-tillegget om medisinsk utstyr her. https://lovdata.no/static/NLX3/32017r0745.pdf
[40] https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2019_11_guidance_qualification_classification_software_en_0.pdf
[41] Håndteringsforskriften: Forskrift om håndtering av medisinsk utstyr - Lovdata
[42] Systemer kategoriseres som høyrisiko-systemer hvis: 1) det inngår som en sikkerhetskomponent i et produkt eller systemet selv er et produkt som reguleres av regelverket opplistet i Annex I og 2) må undergå tredjepartssertifisering etter regelverket i Annex I i KI-forordningen. Artikkel 6.1 i KI-forordningen: Classification rules for high-risk AI systems (eur-lex.europa.eu)
[43] Artikkel 6.2 i KI-forordningen: L_202401689EN.000101.fmx.xml (europa.eu)
[44] Punkt 5 b og c i Vedlegg III i KI-forordningen
[45] Artikkel 27 i KI-forordningen
[46] Artikkel 50 i KI-forordningen
[48] https://single-market-economy.ec.europa.eu/single-market/goods/new-legislative-framework_en
[49] Direktoratet for medisinske produkter (DMP) er fag- og tilsynsmyndighet for medisinsk utstyr i Norge og forvalter produktregelverket for medisinsk utstyr.[1] Utfyllende informasjon finnes på DMP’s nettsider.
[50] Klassifisering - Direktoratet for medisinske produkter (dmp.no)
[51] mdr--oversikt-klassifiseringsregler.pdf (dmp.no)
[52] https://www.dmp.no/globalassets/documents/medisinsk-utstyr/figur-klassifisering---mdr---ny-versjon.png
[53] Se MDR og IVDR artikkel 16
[54] KI-modeller utviklet for bruk på voksne kan ikke uten videre tas i bruk på barn. Se Use of Artificial Intelligence in Radiology: Impact on Pediatric Patients, a White Paper From the ACR Pediatric AI Workgroup (jacr.org)
[55] https://www.dmp.no/medisinsk-utstyr/for-helsetjenesten/egentilvirkning-av-medisinsk-utstyr-i-helseinstitusjoner-in-house
[56] Forordning om medisinsk utstyr (MDR) trådte i kraft 26. mai 2021 og opphevet direktiv 93/42/EØF om øvrig medisinsk utstyr og direktiv 90/385/EØF om aktivt implanterbart medisinsk utstyr med visse unntak. Forordning om in vitro-diagnostisk medisinsk utstyr (IVDR) trådte i kraft 26. mai 2022 og opphevet direktiv 98/79/EF om in vitro-diagnostisk medisinsk utstyr.
[57] Les mer om overgangsordninger på DMPs nettsider.
[58] Søknadspliktige kliniske utprøvinger - Direktoratet for medisinske produkter (dmp.no)
[59] U.S. Food and Drug Administration (fda.gov)
[61] Artikkel 113 i KI-forordningen
[64] Kategoriene beskrives i hhv. KI-forordningens vedlegg I og III. EU-kommisjonen har myndighet til å endre listen over regulerte bruksformål i vedlegg III etter nærmere angitte kriterier, som angitt i artikkel 7 i KI-forordningen.
[65] Artikkel 113 i KI-forordningen samt https://artificialintelligenceact.eu/annex/3/
[67] KI-forordningens artikkel 26 (2): L_202401689EN.000101.fmx.xml (eur-lex.europa.eu)
[68] KI-forordningens artikkel 50(1)(2): L_202401689EN.000101.fmx.xml (eur-lex.europa.eu)
[69] KI-forordningens artikkel 3 (3) definerer ‘general-purpose AI model’ slik: means an AI model, including where such an AI model is trained with a large amount of data using self-supervision at scale, that displays significant generality and is capable of competently performing a wide range of distinct tasks regardless of the way the model is placed on the market and that can be integrated into a variety of downstream systems or applications, except AI models that are used for research, development or prototyping activities before they are placed on the market;
[70] KI-forordningens kapittel V: L_202401689EN.000101.fmx.xml (eur-lex.europa.eu)
[71] KI-forordningens artikkel 53: L_202401689EN.000101.fmx.xml (eur-lex.europa.eu)
[72] KI-forordningens artikkel 55: L_202401689EN.000101.fmx.xml (eur-lex.europa.eu)
[73] https://digital-strategy.ec.europa.eu/en/library/first-draft-general-purpose-ai-code-practice-published-written-independent-experts
[74] https://www.regjeringen.no/no/sub/eos-notatbasen/notatene/2022/des/ai-ansvarsdirektivet/id2950178/
[76] https://www.datatilsynet.no/rettigheter-og-plikter/den-registrertes-rettigheter/rettar-ved-automatiserte-avgjerder/
[77] Personvernforordningen: https://lovdata.no/dokument/NL/lov/2018-06-15-38/KAPITTEL_gdpr-3-2#KAPITTEL_gdpr-3-2
[78] Les mer om dette på Helsedirektoratets nettsider om kunstig intelligens: Regelverket for utvikling av kunstig intelligens - Helsedirektoratet
[79] Dataansvarlig er definert i personvernforordningen som en fysisk eller juridisk person, en offentlig myndighet, en institusjon eller ethvert annet organ som alene eller sammen med andre bestemmer formålet med behandlingen av personopplysninger og hvilke midler som skal benyttes eller det er fastsatt i unionsretten eller nasjonal rett, jf. artikkel 4 nr. 7.Se også pasientjournalloven § 2 bokstav e som viser til definisjonen i personvernforordningen når det gjelder hvem som er ansvarlig for behandling av helseopplysninger i behandlingsrettede helseregistre, herunder i pasientjournaler.
[80] https://lovdata.no/dokument/SF/forskrift/2016-12-16-1659