Lavgradig gliom
Lavgradige gliomer (LGG) er fellesnavn på svulster utgående fra gliaceller med malignitetsgrad 1 og 2 etter WHO-klassifiseringen (Louis et al., 2007). Hos barn er nesten alle disse astrocytomer. LGG skiller seg fra høymaligne svulster ved at tumorveksten hos de fleste er langsom, og hos noen kan endog veksten stoppe spontant uten behandling. Forløpet kan være fluktuerende med periodevis økning og regresjon av tumorstørrelse, hos andre ses et mer aggressivt forløp og endog metastasering til hjernehinner. Som følge av LGGs natur har pasientene en meget høy overlevelse (OS, Overall Survival), mens progresjonsfri overlevelse (PFS) er mye lavere, dvs at progresjon er vanlig, men lar seg oftest vellykket behandle. Barn under ett år har tendens til mer negativt forløp enn eldre barn. Forskjellige histopatologiske undergrupper av LGG har også forskjellig prognose. Svulstenes lavgradige natur er en meget viktig faktor ved vurdering av behandlingsvalg.
Det er nylig publisert europeiske retningslinjer for diagnose og behandling av LGG hos barn og ungdom (A. K. Gnekow et al., 2019).
Epidemiologi
LGG er de vanligste CNS-svulster hos barn og utgjør ca. 40 % av disse. Pilocytisk astrocytom (PA) er den hyppigst forekommende svulst av alle typer registrert i barnekreftregisteret, ca. 10 % av alle pasienter totalt registrert (Stiller, 2007).
LGG er sterkt assosiert med nevrofibromatose 1 (NF-1), og NF-1-pasienter har særlig overhyppighet av pilocytiske astrocytomer i synsveiene (A.K. Gnekow, Packer, & Kortmann, 2004; Stokland et al., 2010). Ofte er disse multifokale med involvering av hjernestammen og andre strukturer.
Histopatologiske typer og lokalisering
Den helt dominerende typen LGG hos barn er pilocytiske astrocytomer (PA) (WHO grad 1) som utgjør over 80 % av gruppen(Stokland et al., 2010). Diffuse astrocytomer grad 2 (DA) er vanligst hos voksne, men utgjør knapt 10 % hos barn, noe hyppigere hos ungdommer. En variant av PA, pilomyxoid astrocytom (PMA) (WHO grad 2) har et mer aggressivt forløp.
Oligodendrogliomer er svært sjeldne hos barn. Diffuse ponsgliomer inkluderes ikke i LGG selv om enkelte av disse histololgisk er DA (WHO grad 2).
Blandingssvulster av nevronale og gliale celler er sjeldne. De er nesten alltid histologisk lavgradige og omtales oftest sammen med LGG. Den vanligste typen er gangliogliom.
LGG kan oppstå i alle deler av CNS, mest hyppig i cerebellum, synsveiene (n. opticus, chiasma, sentrale synsbaner) og hemisfærene (Stokland et al., 2010). Lokalisasjon varierer med alder. Små barn har overvekt av synsveigliomer mens større barn tenderer mot hemisfærebeliggenhet. Pilocytiske astrocytomer er den dominerende typen i alle aldersgrupper, mens fibrillære er noe hyppigere hos ungdommer enn hos små barn. I synsveier og cerebellum finnes nesten utelukkende PA, mens DA utgjør ca. 10–15 % i andre lokalisasjoner.
Molekylærbiologi
De senere år har en funnet svært viktige forandringer molekylærbiologisk hos barn med LGG (Packer et al., 2017). Det gjelder særlig i signalveien MAPK (RAS/RAF/MEK/ERK) som er viktig for cellenes proliferasjon. Spesifikke endringer i denne signalveien kan gi overaktivitet og dermed økt onkogen stimulering. Pilocytisk astrocytoms biologi regnes i dag å representere en «singel pathway disease». Fusjon av BRAF-genet med annet gen (BRAF-KIAA1549) er vanligst ved PA, mens punktmutasjon av BRAF (BRAF V600E) er sjeldnere og ses ved forskjellige LGG. Det foreligger allerede i dag medikamenter som har LGG med slike mutasjoner som mål (MEK-inhibitorer og BRAF-inhibitorer). Se senere.
Diagnose og utredning
LGG gir symptomer enten pga lokal påvirkning eller pga økt intrakranielt trykk, og bildet vil variere sterkt avhengig av lokalisasjon. Synsfunksjonen er ofte påvirket ved supratentorielle midtlinjesvulster og n. opticus-gliomer. Balanseforstyrrelser ses ved cerebellare svulster, epilepsi ved hemisfærelokalisasjon etc. Økt intrakranielt trykk gir hodepine mest uttalt om morgenen, morgenkvalme og brekninger og noen ganger dobbeltsyn (abducensparese). Hvis symptomene har vart lenge er det tegn på lavgradig svulst, men kort varighet er ikke uvanlig.
MR cerebrum viser tumor. På tross av histopatologisk lavgradighet, kan LGG være disseminert eller multifokal, og derfor skal hele CNS visualiseres. Anbefalte MR-teknikker er som ved andre hjernesvulster hos barn.
Histologisk diagnose er oftest nødvendig, og når radikal reseksjon ikke er mulig, gjøres biopsi. Unntak er synsveigliomer med klassiske MR-funn, særlig hos nevrofibromatose-pasienter, der man godtar radiologisk diagnose uten biopsi, fordi disse svulstene erfaringsmessig alltid er PA. Ved tectum-lokalisasjon vil en også være tilbakeholdende med biopsi.
Molekylærbiologisk utredning er ønskelig ved de fleste LGG både for diagnose og valg av behandling.
Oftalmologisk utredning er svært viktig ved supratentorielle midtlinjegliomer, spesielt i synsveiene. Endokrinologisk kartlegging er vesentlig avhengig av lokalisasjon.
Behandling
Behandling og prognose vil variere med lokalisasjon. Laterale svulster vil ligge bedre til rette for radikal kirurgi, og dermed ha bedre prognose. Midtlinjesvulster vil ikke kunne fjernes radikalt og trenger derfor i mange tilfeller ytterligere behandling, ut fra klare retningslinjer (Perilongo, 2005; Reddy & Packer, 1999).
Kirurgi er primærbehandlingen ved LGG (B. J. Due-Tønnessen et al., 2002). Reseksjonsgrad er den viktigste prognostiske faktor, og radikal reseksjon gir best prognose. Ofte vil radikal kirurgi ikke være mulig pga faren for skade, og da synes ikke partiell reseksjon å gi resultatmessige fordeler vedrørende overlevelse sammenlignet med biopsi (Stokland et al., 2010). Men i en del tilfeller vil partiell reseksjon være hensiktsmessig for å bedre pasientens kliniske tilstand, f.eks. ved hydrocephalus. Tilnærmingen må ses i forhold til mulige skadelige effekter av en partiell reseksjon.
Norge har deltatt i den store internasjonale behandlingsprotokollen SIOP-LGG 2004, som ble avsluttet i 2015. En ny studie (LOGGIC) er nå (2019) på det nærmeste ferdigutformet av SIOP-Europe med randomisering i tre behandlingsarmer. Kombinasjonen Vinkristin/Karboplatin er standardarmen, Vinblastin monoterapi som er mye brukt i dag er andre arm, og den mest eksperimentelle armen vil være MEK-inhibitor (Trametinib). Trametinib gis peroralt. LOGGIC åpner i Norge i 2020. NF1-pasienter inkluderes sannsynligvis ikke i denne fordi denne gruppen skal rekrutteres til en annen studie. Ca. 1/3 av alle LGG-pasienter forventes å trenge ikke-kirurgisk behandling. Det er viktig at klare kriterier oppfylles før slik behandling startes. Dette innebærer at ved diagnose / rett etter primærkirurgi, gis medikamentell behandling bare hvis pasienten har alvorlige nevrologiske symptomer som kan forverres, eller truende synstap. Alle andre pasienter blir observert klinisk og billedmessig. Disse pasientene, som utgjør et flertall, er observasjonsgruppen i studien. Dersom tumor i observasjonstiden vokser signifikant ved MR-undersøkelse, eller det er økende symptomer, vurderes da på ny evt kirurgi. Dersom dette ikke er hensiktsmessig, inkluderes også disse pasientene.
Da også de behandlingstrengende pasientene har god prognose vedrørende overlevelse, innfører LOGGIC nye primære endepunkter, særlig gjelder dette synsfunksjonen. Effektiv behandling som bevarer synet vil være sterkt ønsket. Synsundersøkelse med analyse av funksjonsendring vil derfor være en hovedfaktor i studien.
LGG-pasienter med nevrofibromatose (NF-1) planlegges inkludert i et felles studium med USA. Det ligner LOGGIC, men har bare to armer: vinkristin/karboplatin vs. MEK-inhibitor. Dersom studien ikke lar seg gjennomføre, vil gruppen bli inkludert i LOGGIC.
Strålebehandling kan ha god effekt ved LGG og var tidligere mye brukt. Pga mulige kognitive seineffekter og sekundærkreft, er radioterapi nå ikke en del av primærbehandlingen. Imidlertid kan det være aktuelt når medikamentell behandling ikke er effektiv nok, spesielt ved små svulster og hos eldre barn og ungdom. Strålekniv kan brukes i utvalgte tilfeller. Hos NF-1-pasienter vil en være enda mer tilbakeholdende med strålebehandling pga økt følsomhet for alvorlige strålebivirkninger
En oppnår sjelden komplett remisjon av medikamentell behandling, men varig stabil tumorstørrelse eller noe reduksjon vurderes som tilfredsstillende behandlingseffekt.
Anbefalinger:
- Kirurgi anbefales når radikal tumorreseksjon er mulig, og denne behandlingsmodus skal alltid vurderes først.
- Når radikal kirurgi ikke er mulig, skal biopsi gjøres i de aller fleste tilfellene. Partiell reseksjon synes ikke å gi bedre overlevelsesresultater enn biopsi og er derfor omdiskutert. Dette må tas stilling til i hvert enkelt tilfelle.
- Kirurgi vurderes på ny ved residiv eller progresjon.
- Når klare kriterier er oppfylt (se ovenfor), anbefales å gi nonkirurgisk behandling i henhold til LOGGIC protokollen.
- Pasienter med nevrofibromatose behandles etter egen protokoll.
Ny utprøvende behandling
MEK-inhibitor hemmer MAPK-signalveien «downstream» for BRAF og har vist seg å gi god respons ved LGG i ferske studier. Dette har ført til at trametinib nå skal brukes «up-front» i LOGGIC-studien. Langtidseffekten er foreløpig ikke kjent, men ved behandlingsrefraktær LGG med dårlig prognose bruker mange nå MEK-inhibitor, peroralt en gang daglig. En forventer etter hvert mer kunnskap om virkninger på lengre sikt.
BRAF-inhibitor gir god respons på svulster som har BRAF-punktmutasjon, og er i dag førstevalg når slik mutasjon er påvist. Imidlertid gir den paradoksal effekt i form av økt tumoraggressivitet ved svulster med BRAF-fusjon, og må derfor aldri gis uten bekreftet BRAF V600E punktmutasjon.
Bevacizumab alene eller kombinert med andre midler brukes ved resistent LGG i dag. Den gir god respons hos tidligere sterkt behandlede pasienter, men nesten alle får progresjon etter avsluttet behandling(Hwang et al., 2013). Positiv påvirkning av visus har vært rapportert, men flere studier har ikke kunnet verifisere dette.
Behandling av tilbakefall eller progresjon etter primærbehandling
LGG har et helt annet forløpsmønster enn aggressive kreftformer. Ofte starter tumor ny vekst flere år etter at den har blitt stabilisert av behandling. En kan da gi samme behandling som initialt, eller andrelinjebehandling. I Norge er det for tiden vanligvis vinblastin monoterapi. MEK-inhibitor kan vurderes som anført ovenfor.
Anbefalinger:
- Dersom primærbehandlingen hadde god effekt, kan en gjenta denne.
- Ved tidlig progresjon anbefaler vi annenlinjebehandling. Mest brukt nå er ukentlig vinblastin i ett år (Bouffet et al., 2012).
Organisering av behandlingen
Behandlingen av LGG er regionalisert og ledes av det regionale barnekreftsenteret. Dette gjelder både kirurgi, kjemoterapi og strålebehandling. Deler av kjemoterapien kan gis mer lokalt.
Prognose
Mortaliteten er lav med 5 års overlevelse rundt 95 %, mens progresjonsfri overlevelse (PFS) er atskillig lavere. Komplett reseksjon er den viktigste uavhengige positive prognostiske faktoren, mens alder under ett år ved diagnose og de viktigste grad 2-histopatologiske typene (DA og PMA) er negative faktorer. Ved svulster i synsveiene har NF1-pasienter bedre prognose enn andre (Stokland et al., 2010). Ved synsveigliomer får pasientene ofte senskade i form av betydelig og varig synsreduksjon. DA kan ha en tendens til å utvikle seg til malignt astrocytom etter flere år, men i langt mindre grad enn hos voksne. Dette skjer svært sjelden ved PA, men er beskrevet.
Oppfølging og seineffekter Anbefalinger:
- Klinisk kontroll med MR gjøres hver tredje måned de første to år, deretter hver 6. måned, og deretter årlig.
- Progresjon kan oppstå etter flere år, slik at oppfølging i ca. 10 år anbefales.
- Oftalmologisk oppfølging er essensielt hos pasienter med synspåvirkning.
- Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose ved regionsykehusene i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri, og det gjøres en vurdering av behov for utredning også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig.
- Habilitering og andre former for oppfølging individualiseres.
Høygradige gliomer og Diffuse ponsgliomer
Høygradige gliomer (HGG) er de vanligste CNS-svulster hos voksne, men betydelig sjeldnere hos barn. De aller fleste er astrocytomer, og etter oppdatert WHO- klassifiseringen av CNS svulster fra 2016 skilles det mellom anaplastisk astrocytoma IDH wild type eller IDH mutert (AA) (WHO grad 3), glioblastoma (GB) IDH wild type eller IDH mutert (WHO grad 4) og diffuse midtlinje gliomer, H3K27M-mutert (WHO grad 4) (Gupta & Dwivedi, 2017). Høygradige astrocytomer hos barn har en annen mutasjonsspektrum sammenlignet med voksne svulster. For eksempel viser anaplastiske astrocytomer hos barn veldig sjelden IDH mutasjon som er mer typisk hos voksne. Diffuse ponsgliomer (DIPG- Diffuse Intrinsic Pontine Glioma) er en undergruppe av diffuse midtlinje gliomer og omtales separat da de har egen behandling på grunn av spesifikk biologi og klinisk forløp med svært dårlig prognose.
Diffuse/Fibrillære astrocytomer (WHO grad 2) er lavgradige gliomer, men det kan være en vanskelig avgrensning mot grad 3, og svulstene kan utvikles i malign retning med tiden.
Vi finner HGG i hele CNS, fra 35–50 % supratentorelt, 40–50 % oppstår i hjernestammen inkludert ponsområdet som diffuse ponsgliomer(DIPG), i midtlinjestrukturer inkludert thalamus (13 %), i lillehjernen (5 %) og i spinalkanalen (3 %) (Gianno F, 2018).
Epidemiologi
HGG har en insidens på ca. 0,85 pr 100 000 barn og ungdom fra 0–19 år pr år og utgjør ca. 8–12 % av alle CNS-svulster hos barn (Braunstein, Raleigh, Bindra, Mueller, & Haas-Kogan, 2017).
Strålebehandling mot CNS fører på sikt til en økt risiko for utvikling av HGG.
Patologi
Både AA og GB vokser diffust infiltrativt og tumor kan være vanskelig å avgrense på MR bilder. Begge svulstene har høy mitotisk aktivitet, høy cellularitet, kjerneatypi og cellulær pleomorfisme. Hos GB finner vi i tillegg varierende områder med nekrose, småblødninger, fast og bløtt vev, i tillegg til mikrovaskulær proliferasjon.
Skillet mellom grad 3 (AA) og grad 2 (DA/FA) kan være vanskelig, spesielt i små biopsier, og patologens diagnostisering vil influere betydelig på resultatene i studier for hhv HGG og LGG etter hvordan grensetilfellene blir vurdert.
Molekylærbiologi
De siste årene har det vært en voldsom økning i kunnskapen om molekylærbiologiske forhold ved hjernesvulster og for HGG er det funnet stor heterogenitet. Kartlegging av arvematerialet ved HGG har bl.a. påvist mutasjoner som affiserer histonfunksjonen i svulstcellene (K27M og G34R/V mutasjon i H3.3 og H3.1), og som er et kjennetegn ved HGG hos barn og unge voksne. Videre er det funnet molekylære subgrupper (for.eks. H3.3 K27M, H3.1 K27M, H3.3 G34R/V) som kan si noe om svulstens plassering i CNS, typisk alder når svulsten opptrer og om prognose.
I kortikale svulster finnes ofte mutasjon eller overekspresjon av TP53, i tillegg til metylert MGMT og 5–10 % av svulstene i dette område utrykker BRAF V600E mutasjon.
Det finnes også andre genetiske forandringer som epidermal vekstfaktor-reseptor (EGFR) og platederivert vekstfaktor-reseptor A (PDGFRA) som kan være mutert og/eller ha amplifikasjon/overekspresjon i ulike sub-grupper av HGG (Sturm, Pfister, & Jones, 2017).
Diagnose og utredning
Som ved de fleste andre hjernesvulster avhenger symptomene ikke av histologi, men mest av tumors lokalisasjon som forårsaker lokal påvirkning eller økt intrakranielt trykk. Ofte har symptomene vart i kort tid pga. svulstenes aggressive vekst. Hodepine, kvalme/oppkast, epilepsi og hemiplegi er vanlige symptomer. Andre fokalt fremkalte symptomer forekommer. Rask forverring skjer hvis pasienten ikke blir behandlet.
Ved MR undersøkelse av HGG er tumor uklart begrenset med irregulære marginer. Som oftest ses inhomogen kontrastoppladning. GB viser oftest sentral nekrose omgitt av en hyperintens sone og ødem rundt tumor er vanlig.
Tumor er som oftest fokal, men kan gi spinal spredning, slik at hele CNS skal visualiseres.
Histopatologisk diagnose er helt nødvendig, og materiale fås ved reseksjon eller biopsi.
Behandling
Kirurgi er en svært viktig del av behandlingen, og målet er å fjerne hele svulsten radikalt hvis mulig, eventuelt så mye som mulig uten for stor skade. Da svulsten har et infiltrativt vekstmønster, vil det alltid være gjenværende tumorceller rundt reseksjonen.
Fokal strålebehandling gis til alle barn over 3 år. HGG har ikke mikrospredning til CNS slik at stråling av hele CNS er ikke nødvendig. Senskadene blir dermed mindre.
Kjemoterapi
Allerede i 1976 ble det i USA startet behandling med kjemoterapi bestående av prednison, lomustin og vincristin (pCV), med signifikant bedring av resultatene i forhold til stråling alene. Det samme ble vist i andre studier hvor kjemoterapi i tillegg til strålebehandling gav økt overlevelse i forhold til bare stråling (Sposto et al., 1989). Senere har det i flere studier vært ny histologisk gjennomgan av tumorvev som har vist at mange lavgradige gliomer har vært inkludert i studiene, og at kjemoterapi i tillegg til kirugi og stråling ikke gav bedre overlevelse ved HGG (Fouladi et al., 2003).
Senere utprøving av nye regimer har ikke klart å bedre resultatene for HGG. Temozolomid sammen med og etter strålebehandling har hatt en viss effekt hos voksne, men det er ikke funnet samme effekt hos barn(K. J. Cohen et al., 2011).
Det prøves ut en rekke nye stoffer og prinsipper i behandlingen av HGG hos barn. Lovende resultater har blitt oppnådd med vaksinasjon med dendritiske celler. Ut fra økende molekylær forståelse av HGG hos barn prøves det ut stoffer som påvirker forskjellige molekylære signalveier, i form av monoklonale antistoffer, småmolekylinhibitorer, strålesensitizere, genterapi og immunterapi. Disse kan påvirke celleproliferasjon, angiogenese, invasivitet og celleoverlevelse (Hargrave, 2009). Rebestråling av supratentoriale HGG har vist forlenget levetid sammenliknet med barn som ikke fikk rebestråling (Tsang, Oliveira, et al., 2019).
For øyeblikket er det ingen åpne studieprotokoller for barn med HGG. Internasjonalt er det ikke konsensus om beste behandling. De fleste barn i Norge får kombinasjonsbehandlingen med stråling og Temozolomid eller stråling og to ulike kjemoterapi, Temozolomid og Lomustine. Den siste kombinasjonen har vist seg å være mer effektiv enn Temozolomid alene, spesielt hvis tumorvevet utrykkeraktiv (umetylert) MGMT enzym promotor som medfører begrenset effekt av Temodal (Jakacki et al., 2016).
HGG hos barn < 3 år er veldig sjeldent og de har en bedre prognose sammenliknet med eldre barn, grunnet ulik biologi i svulstvevet (Gielen et al., 2015). Disse barna har nytte av kjemoterapi og anbefales behandling etter «SIOP Infant HGG protocol».
Prognose
Det har vært lite fremgang i resultatene for HGG. 5 års overlevelse ligger mellom 15 og 25 %. Histologi og omfanget av tumorreseksjon er viktige prognostiske faktorer, i tillegg til proliferasjonsindex. AA har bedre prognose enn GB og total tumorreseksjon og lavere proliferasjons index gir bedrer prognosen. I omtrent 40 % av HGG påvises mutasjon i tumor supressor genet, TP53, noe som er forbundet med dårlig prognose. Påvisning av mutasjoner i H3F3A, genet som koder for histonfunksjonen i svulstcellene, gir opphav til ulike molekylærbiologiske subgrupper som kan si noe om svulstens plassering i CNS, typisk alder når svulsten opptrer og om prognose. Svulster med H3.3 K27M mutasjon og H3.1K27M mutasjon finnes ofte i hjernestammen og har svært dårligprognose, mens H3.3 G34R/V mutasjon er cortikal svulster med bedre prognose (Braunstein et al., 2017).
BRAF V600E mutasjon finnes i 5–10 % ab HGG og er forbundet med bedre prognose. For disse pasientene finnes det «target therapy» i form av BRAF inhibitor som så langt har vist lovende resultater (Robinson, Orr, & Gajjar, 2014).
Diffuse ponsgliomer
Dette er gliomer som infiltrerer diffust i den sentrale hjernestamme og utgjør en egen klinisk tilstand, uavhengig av tumors histopatologi, som i de aller fleste tilfeller er astrocytom WHO grad 2, 3 eller 4. På engelsk kalles svulsten «Diffuse Intrinsic Pontine Glioma» (DIPG). Tumor sitter nesten alltid sentralt i pons, og diagnosen stilles på bakgrunn av klassiske MR-funn og spesifikke kliniske symptomer. Prognosen er særs dårlig, bare spredte langtids overlevere er beskrevet. Det er gjort mange behandlingsforsøk med forskjellige tilnærminger uten at noen har hatt tilfredsstillende effekt. Strålebehandling har en viss livsforlengende effekt.
Det er imidlertid viktig å være klar over at det også finnes mer fokale (ikke- diffuse) svulster i hjernestammen med et helt annet forløp. Disse er som oftest lavgradige gliomer med forløp og behandling som LGG andre steder. Prognosen ved LGG i hjernestammen varierer, men kan være ganske god med høy langtidsoverlevelse. (Bemerk at fibrillært astrocytom (WHO grad 2) som manifesterer seg som DIPG ikke regnes som LGG.)
Epidemiologi
Svulster i hjernestammen utgjør 10–15 % av alle pediatriske CNS svulster hos barn og DIPG utgjør 80 % av disse. DIPG forekommer i alle aldersgrupper, men er hyppigst å alderen 5–10 år (Vanan & Eisenstat, 2015).
Patologi
Tidligere var det ikke vanlig å biopsere DIPG, både på grunn av svulstens beliggenhet i et sårbart område og begrenset terapeutiske muligheter. Muligheten for å finne molekylære endringer i svulstvevet og påfølgende målrettet terapi, har ført til en økt biopsering av DIPG, noe som har vist seg å være relativt trygt. Den pågående BIOMEDE studien for DIPG hos barn og ungdommer, krever biopsi for inklusjon i studien.
Nesten alle biopserte svulster viser astrocytom WHO grad 2–4 og fordelingen mellom disse varierer i publiserte materialer, men forløpet varierer ikke mellom de forskjellige histopatologiske typene. Det er beskrevet enkelte DIPG med PNET histopatologi.
Molekylærbiologi
Den økende biopsering ved DIPG har vist at over 80 % –90 % av DIPG innehar H3K27M mutasjon som affiserer histonfunksjon i svulstcellene (Sturm et al., 2017). Dette er bakgrunnen for den nye histologiske diagnosen i siste WHO-utgave (Gupta & Dwivedi, 2017) . Denne mutasjonen finnes også i andre høygradige midtlinjegliomer.
Diagnose og utredning
På grunn av sykdommens aggressive biologi, inntrer symptomene akutt og progredierer raskt. Oftest har symptomene vart mindre enn 2–3 måneder ved diagnose. Pasientene har klassiske hjernestammesymptomer som abducensparese, facialisparese, dysfagi, hemiplegi og ataksi.
MR cerebrum gir diagnosen. Svulsten utvider pons og infiltrerer diffust omgivelsene langs fibertraktene. Den gir betydelig masseeffekt med sammenklemming av fjerde ventrikkel, men mindre enn 10 % har hydrocefalus ved diagnose. Tumor er hypointens på T1, hyperintens på T2 og FLAIR. Den tar sjelden kontrast. Det siste er viktig differensialdiagnostisk i forhold til pilocytisk astrocytom.
DIPG diagnostiseres radiologisk og er alltid inoperabel. Biopsi av DIPG er blitt mye vanligere, grunnet muligheten for å finne molekylære endringer i svulstvevet og påfølgende målrettet terapi,
Ved ikke-diffuse hjernestammesvulster og når det er tvil om diagnosen DPG, skal det gjøres biopsi.
Behandling
Kirurgi. DIPG er alltid inoperabel. Vedrørende biopsi, se ovenfor.
Strålebehandling er standardbehandling og den eneste behandling med dokumentert effekt ved DIPG. Effekten er forbigående klinisk bedring og opptil 3 mnd. forlengelse av levetiden. Vanlig dose er 54 Gy til tumorområdet. Det gis ikke stråling til hjernen for øvrig. Behandlingen er ikke kurativ. Disse pasientene er i øyeblikket ikke kandidater for protonstrålebehandling. Re-bestråling har vist økt overlevelse, sammenliknet med standardbehandling (Lu, Welby, Mahajan, Laack, & Daniels, 2019).
Kjemoterapi og annen behandling. De fleste cytostatika i varierende kombinasjoner og doser har blitt prøvd kombinert med strålebehandling, uten bedre effekt enn strålebehandling alene. Høydosebehandling har heller ikke bedret resultatene, heller ikke Angiocomb, en studie som kombinerte topotecan som radiosensitizer gitt under strålebehandlingen, etterfulgt av kurer med de anti-angiogenetiske medikamentene thalidomid og celecoxib, samt etoposid. Utprøving med stoffer som gemcitabine (nukleosid analog), gefitinib (EGFR inhibitor), tipifarnib (farnesyltransferase inhibitor) og flere tyrosin kinase hemmere, som påvirker molekylære prosesser i svulsten, har heller ikke vist bedre resultater. I Norge (2019) inkluderer vi pasienter i BIOMEDE (Biological Medicine for Diffuse Intrinsic Pontine Glioma (DIPG) Eradication (BIOMEDE), 2014-), som er en europeisk behandlings-protokoll for DIPG. Etter biopsi fra tumor, blir alle bestrålt, i tillegg til biologisk medisin som styres etter svulstens biologiske karakter (Everlimus (mTOR inhibitor), Erlotinib eller dasatinib (begge tyrosin kinase hemmer). Resultatene fra BIOMEDE forventes i løpet av 2020.
Prognose
Prognosen er svært dårlig med median overlevelse på 9–13 måneder. Dette har ikke endret seg de senere år. Overlevelsestall varierer i litteraturen, ofte kan det være uklart om angitte overlevere egentlig har en annen type hjernestammesvulst. Det er imidlertid påvist enkeltpasienter med godt dokumentert DIPG som overlever lenge.
Det er ikke beskrevet prognostiske faktorer som har behandlingsmessige konsekvenser, men kort sykdomsvarighet og betydelige nevrologi ske symptomer har en viss negativ prognostisk betydning. Lavere malignitetsgrad (WHO grad 2) har ikke innflytelse på forløpet.
Ependymom
Ependymom er den tredje vanligste hjernesvulsten hos barn og utgjør 8–12 % av alle hjernesvulstene hos barn og ungdom. Mer enn halvparten av tilfellene opptrer hos barn under 5 år og vi antar at svulsten utgår fra ependymcellene som dekker hjernens ventrikler og sentralkanal i spinalkanalen (Khatua, Ramaswamy, & Bouffet, 2017).
Lokalisering
Ependymomene er stort sett lokalisert i tilknytning til ventriklene, men kan forekomme i hele sentralnervesystemet (CNS), uavhengig av og langt fra områder med ependymceller. Det forklares ved transformasjon av embryologiske rester av ependymceller inne i hjerneparenchymet eller en transformasjon av nevrale stamceller (Reni, Gatta, Mazza, & Vecht, 2007; Zacharoulis & Moreno, 2009). Hos barn er 90 % av ependymomene intrakranielle og av disse er 2/3 lokalisert i bakre skallegrop i tilknytning til 4. ventrikkel (Khatua et al., 2017).
Histopatologi og molekylærbiologi
Etter oppdatert WHO- klassifiseringen av CNS svulster fra 2016 skilles det mellom WHO grad I subependymom og myksopapillær ependymom, som hovedsakelig opptrer hos voksen, WHO grad II ependymom og WHO grad III anaplastisk ependymom (Gupta & Dwivedi, 2017). For WHO grad II ependymomer finnes tre histologiske varianter: pappillært, klarcellet og tancytisk ependymom. RELA fusjon positiv ependymom er en molekylær variant av supratentorielle ependymomer og kan enten være WHO grad II eller WHO grad III. De vanligste histopatologiske subtypene hos barn er klassisk og anaplastisk ependymom. Den klassiske typen viser perivaskulære pseudorosetter av gliale tumorceller uten tydelig tegn til anaplasi, mens den anaplastiske typen viser forhøyet mitotisk aktivitet, mikrovaskulær proliferasjon og utbredte nekroser. Det er glidende overganger mellom de to typene og ofte vanskelig å avgjøre hvilken gruppe de tilhører (Ellison et al., 2011). Det er foreløpig ingen pålitelige biologiske prognostiske markører for ependymom, og sammenhengen mellom det histopatologiske bildet og prognose har ikke vist seg å gjenspeile seg i kliniske studier (Ellison et al., 2011; Lundar, Due-Tonnessen, Scheie, & Brandal, 2014).
De siste årenes økning i kunnskapen om molekylærbiologiske forhold ved hjernesvulster har vist en stor heterogenitet når det gjelder ependymom og det er funnet 9 molekylære subgrupper av ependymale svulster, 3 for hvert hovedområde i hjernen: supratentorielt, bakre skallegrop og spinakanal. For supratentoriale ependymom (ST-EPN) hos barn er anaplastisk EPN med YAP1-fusjon og anaplastisk EPN med RELA-fusjon de vanlige subgruppene, hvor YAP1-fusjon er forbundet med svært god prognose og RELA-fusjon langt dårligere prognose. For ependymom i bakre skallegrop (PF-EPN) er det også hovedsakelig 2 subgrupper hos barn, anaplastisk gruppe A med dårlig prognose og anaplastisk gruppe B med god prognose. I spinalkanalen er det funnet 3 subgrupper, men alle er forholdsvis sjeldne hos barn (Pajtler et al., 2015). Forekomsten av tillegg av kromosom 1q indikerer dårlig prognose og stor sannsynlighet for tilbakefall.
Diagnose og utredning
De vanligste symptomene ved ependymom er spesifikke og relatert til økt intrakranielt trykk fra obstruktiv hydrocephalus som morgenhodepine, brekninger om morgenen og evn. personlighetsforandringer, men nevrologiske utfall og cerebellare symptomer som ataksi kan forekomme. Hos små barn kan apati og irritabilitet være de eneste symptomene. Diagnosen stilles ved MR-undersøkelse av hele CNS-aksen, undersøkelse av spinalvæsken og histologisk undersøkelse av svulstvevet.
Behandling
Behandling av ependymom er som oftest multimodal og krever høyspesialiserte, bredt sammensatte nevro-onkologiske team. Den gjennomføres i henhold til internasjonale studieprotokoller/retningslinjer.
Kirurgi
Standard behandling av ependymom er først og fremst kirurgi, etterfulgt av fokal strålebehandling (Merchant et al., 2009). Kjemoterapi har en mer usikker effekt på svulstvevet (Bouffet, Tabori, Huang, & Bartels, 2009). Den optimale behandlingen vil være total tumorfjerning, men ofte er det vanskelig grunnet svulstens evne til å infiltrere vitale strukturer, spesielt i bakre skallegrop. Målet for kirurgisk behandling vil være maksimal tumorfjerning, samtidig med et optimalt nevrologisk resultat. Studier har vist at det som betyr mest for overlevelsen, er grad av resttumor. Det har den senere tid ført til en mer aggressiv kirurgisk tilnærming med «second look» kirurgi for å oppnå totalreseksjon av tumor, og dermed bedre overlevelsen (Merchant et al., 2009). Studier har vist at det er gjennomførbart uten alvorlig morbiditet (Massimino et al., 2004).
Strålebehandling
Standard behandling av ependymom er kirurgi, etterfulgt av strålebehandling hos større barn og ungdom og kjemoterapi hos de minste barna som ikke kan få strålebehandling. Studier har vist at behandling med både kirurgi og strålebehandling har bedre overlevelse (Schroeder et al., 2008; Swanson et al., 2011) Protonstråling bør være standard behandling.
CNS-metastaser forekommer, men i langt mindre frekvens enn man tidligere trodde og derfor er det kun tumorområdet med marginer som blir bestrålt. Dosen må over 54 Gy og helst så høyt som 59.4. I den nyeste behandlingsprotokollen SIOP Ependymoma II er stråledosen 59,4 Gy for barn > 18 mnd., med mindre stråledoser (54 Gy) til barn mellom 12–18 mnd. og til barn som har gjennomgått flere kirurgiske inngrep (An International Clinical Program for the Diagnosis and Treatment of Children With Ependymoma (SIOP-EP-II), 2015-2026).
For ependymom i hjernestammen er det flere land, inkludert Norge, som har vært skeptisk til en så høy stråledose (59,4 Gy) mot et sårbart område og påfølgende fare for strålenekrose.
Cytostatika
Cytostatikabehandling ved ependymom har usikker effekt og beste standard behandling er ikke etablert (Bouffet et al., 2009). Cytostatika brukes på de minste barna i påvente av strålebehandling og cytostatika har trolig en viss effekt (Grundy et al., 2007). Andre studier viser ikke den samme effekten (Grill et al., 2001).
I «SIOP Ependymoma II» får alle spedbarn < 12 måneder og barn med resttumor, cytostatika. I protokollen inngår cyclofosfamid, vincristin, carboplatin, etoposid, cicplatin og metotrexate i forskjellige kombinasjoner. I tillegg blir spedbarn < 1 år eller barn som ikke har fått strålebehandling, randomisert til Valporat eller ikke i vedlikeholdsbehandlingen.
Barn med ependymom i Norge behandles i utgangspunktet etter «SIOP Ependymoma II», men med visse modifikasjoner. Stort sett vil en i Norge gå for mindre stråling enn anbefalt i protokollen når tumor ligger i hjernestammen. Faggruppen for CNS svulster hos barn har bestemt at Norge skal delta i «SIOP Ependymoma II», og arbeidet med å åpne studien i Norge er allerede i gang.
Prognose
Prognosen for ependymom er relativt dårlig med 5 års PFS mellom 23–74 % og OS mellom 60 %–80 % (Cage et al., 2013). Den varierer med alder, WHO-klassifikasjon, lokalisasjon og resttumor etter kirurgi. Det som har betydd mest prognostisk er grad av resttumor etter kirurgi og ved komplett reseksjon av WHO grad 1–3 er det > 60 % 5-års overlevelse (Pajtler et al., 2017; Pejavar et al., 2012). Overlevelse basert på de nye molekylære subgruppene av ependymale svulster, viser at ST anaplastisk EPN med YAP1-fusjon og PF anaplastisk EPN gruppe B har svært god prognose (10 års OS 88–100 %), mens ST anaplastisk RELA-fusjon og PF anaplastisk EPN gruppe A har langt dårligere prognose (10 års OS 50 %) (Pajtler et al., 2015).
Oppfølging
Alle barn kontrolleres med klinisk undersøkelse og MR av hele CNS hver 3. mnd. første året etter avsluttet behandling, deretter hver 4. mnd. andre året og videre halvårige kontroller frem til 5 år etter avsluttet behandling. I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. Mange barn med ependymom vil i varierende grad få senfølger av behandlingen. Dette må følges nøye. Nevropsykologisk kartlegging gjennomføres på alle 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingsprogram, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Videre oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus gjøres i varierende grad. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette for best mulig habilitering
Residivbehandling
Residiv ved ependymom forekommer hos 30–50 % av pasientene og er forbundet med veldig dårlig prognose (Zacharoulis et al., 2010). Det finnes ingen standard behandling. Nytt operativt inngrep må alltid vurderes og der hvor strålebehandling ikke er gjennomført, må det vurderes. Kraniospinal re-bestråling har vist økt overlevelse sammenliknet med fokal re-strålebehandling, mens cytostatika har vist dårlig effekt (Tsang, Murray, et al., 2019). Utprøvende behandling med biologisk medisin vil mulig gi større håp for denne pasientgruppen i fremtiden.
Medulloblastom
Medulloblastom er en malign svulst av embryonal opprinnelse lokalisert i lillehjernen, som opptrer særlig hos barn. Det er en biologisk heterogen svulstgruppe, og det er nå bred enighet om at det eksisterer 4 molekylærgenetiske subgrupper med betydning for prognose. I WHOs klassifikasjon av svulster i sentralnervesystemet fra 2016 er disse nå tatt med, i tillegg til de histologiske undergruppene av medulloblastom (Louis et al., 2016).
Symptomene er enten sekundært til svulstens tendens til å gi vannhode/ hydrocephalus eller direkte påvirkning av (lille)hjerne eller ryggmarg. Kvalme, brekninger, trunkal ataxi og koordinasjonsproblemer er de vanligste symptomene.
Diagnose
MR-undersøkelse som fremstiller hjerne og ryggmarg er nødvendig og gir mistanke om diagnosen, men endelig diagnose og klassifisering kan ikke bestemmes før etter histologisk og molekylærbiologisk undersøkelse av svulstmaterialet og spinalvæske.
WHOs klassifikasjon av medulloblastomer (2016)
Medulloblastomer klassifiseres under embryonale tumores og er høygradig maligne (WHO grad IV). Følgende undergrupper er definert:
Både molekylærbiologisk og histologisk definert medulloblastom brukes når diagnosen stilles, og de ulike kombinasjonene av genetiske og histologiske undergrupper har ulik prognose og forekommer i ulike aldersgrupper. Se tabell under:
Table 5: Summary of the most common integrated medulloblastoma diagnoses, with clinical correlates
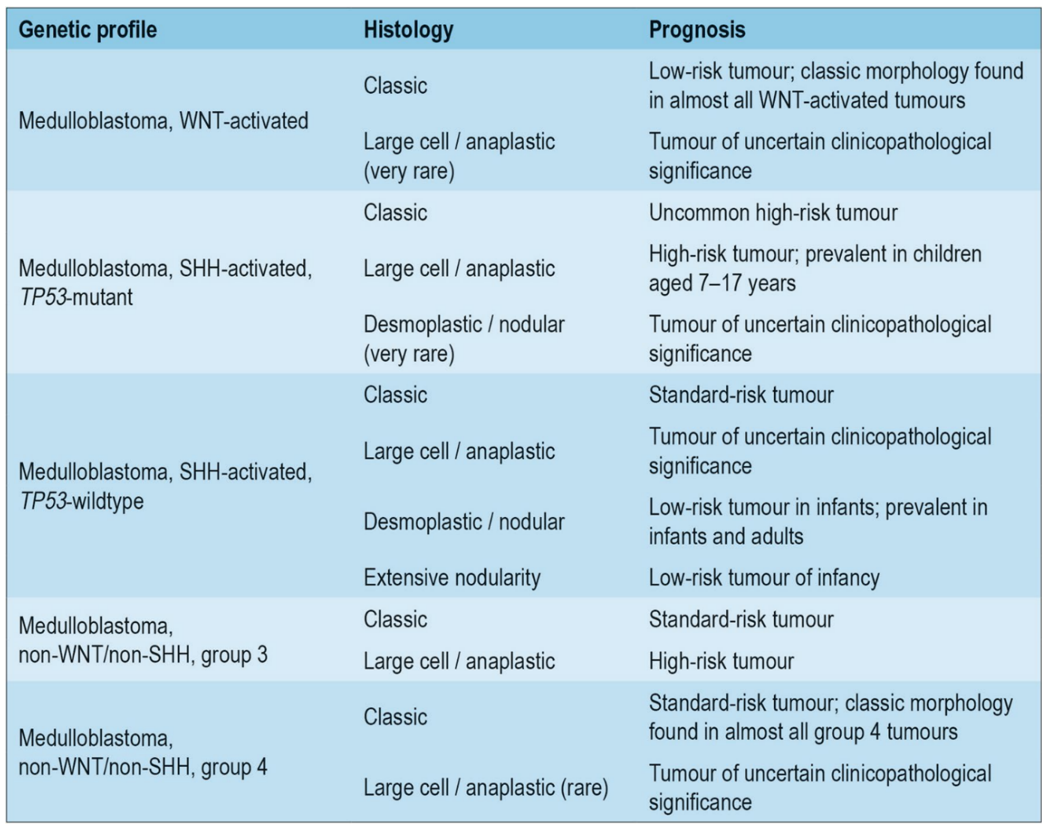
Molekylærbiologiske markører
Tumormateriale vil bli undersøkt på følgende markører for å evaluere prognostisk betydning: CTNNB1-mutasjoner (WNT-aktiverte medulloblastomer), APC-mutasjoner (WNT-aktiverte CTNNB1-villtype medulloblastomer), monosomi 6 (WNT-aktiverte medulloblastomer), gain/amplifikasjon av C-MYC og N-MYC, TP53-mutasjoner, PTCH1-mutasjoner, og SUFU-mutasjoner (SHH-aktiverte medulloblastomer).
Flere av disse markørene er implementert som prognostiske faktorer i behandlingsprotokoller.
Stadieinndeling (Chang)
M0: | Lokalisert til lillehjernen uten tegn til lokal spredning eller spredning til hjernehinnevæsken |
M1: | Spredning til hjernehinnevæsken |
M2: | Spredning lokalt i lillehjernen |
M3: | Spredning til storehjerne/ryggmargen |
M4: | Spredning utenfor sentralnervesystemet |
Standard risk: | M0 og postoperativ resttumor under 1.5 cm2 |
Høyrisk: | M1–4, storcellet eller anaplastisk histologi, og/eller MYC-amplifisert |
Behandling
Kirurgi alene er ikke kurativt ved medulloblastom. Adjuvant behandling er helt nødvendig for overlevelse. Barn over 4 år vil rutinemessig bli behandlet med strålebehandling, helst protonbestråling, mot hjerne og ryggmarg. Barn under 4 år vil bli behandlet med kjemoterapi som eneste adjuvante behandling. Opptil 1/3 av pasientene har spredning til hjernehinnene ved diagnose. Valg av behandlingsprotokoll og -stratifisering vurderes og bestemmes i MDT-møte.
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Ved restsvulst på mindre enn 1,5 cm2 på postoperative MR-bilder ser man en bedre prognose/overlevelse enn ved større resttumor. Som for de fleste andre svulsttyper vil derfor målsetningen med kirurgi ved medulloblastom være maksimal svulstreseksjon uten å løpe for stor risiko for å påføre pasienten nevrologiske sekveler.
Stråleterapi er en viktig del av behandlingen og hele nevralaksen skal bestråles med boost mot primærtumorområdet og eventuelle metastaser. Dose til nevralaksen varierer fra 18 til ca 36 Gy, mens man til primærtumorområdet og eventuelle metastaser totalt gir 54 Gy (ca 50 Gy ved bestråling spinalt). Grunnet risiko for alvorlige senskader anbefales ikke bestråling til de minste barna. Stråling med protoner er standardbehandling i dag. Mer spesifikt gir man for norske barn under 4 år ikke strålebehandling til nevralaksen, mens man vurderer om det er aktuelt med lokal bestråling mot tumortomt og eventuell resttumor for de som er fra 2 år og oppover. Ett av ankepunktene mot sistnevnte strategi er at det ved recidiv blir vanskeligere å gi ny strålebehandling.
Cytostatikabehandling er under kontinuerlig evaluering. Medulloblastomer er vanligvis kjemoterapisensitive, men beste standardbehandling er ikke endelig fastslått. Hos barn < 4 år startes det med postoperativ cytostatikabehandling i håp om å utsette stråleterapi lengst mulig (Rutkowski, 2006; Rutkowski et al., 2005). Hos noen spesifikke undergrupper av medulloblastom hos de aller yngste barna, er det også god prognose ved kun adjuvant behandling med cytostatika, inkludert methotrexate intratekalt.
Standard risk < 4 år: | HIT MED Guidance |
Standard risk > 4 år: | SIOP PNET 5 |
Høyrisk alle aldre: | HIT MED Guidance |
Cytostatika som inngår er cyclofosfamid, vinkristin, metotrexat, karboplatin, etoposid, cisplatin og CCNU i varierende kombinasjoner og intensitet alt etter alder og sykdomsutbredelse. Mange av høyrisk-pasientene og de under 4 år får også intratekal metotrexat. Alle barn over 4 år får bestråling mot CNS-akse. Barn under 4 år behandles rutinemessig med kun cytostatika.
Prognose
Best prognose har standard risk pasienter med ca 70 % langtidsoverlevelse, mens prognosen for de yngste barna (< 4 år) er dårligere. Høyriskpasientene > 4 år har ca 40–50 % langtidsoverlevelse, og også blant disse er det dårligst prognose for de minste barna (< 4 år).
Oppfølging
Alle barn kontrolleres og diskuteres i MDT-møter. Rutinemessig billedoppdatering og tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, siden MR hvert år til 5 år etter avsluttet behandling. Etter 5 år er det individuell oppfølging, men pga at de fleste har fått strålebehandling mot total CNS-akse, vil vi anbefale MR-kontroller livslangt.
I tillegg bør alle barn følges minst 1 gang årlig med tanke på sekveler av behandlingen. Alle barn med medulloblastom vil i varierende grad få senfølger av behandlingen. Nevropsykologisk kartlegging gjennomføres for alle 1, 2 og 5 år etter diagnosetidspunkt i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Videre oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus gjøres i varierende grad. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering.
Residivbehandling
Ved residiv er behandlingsresultatene nedslående. De terapeutiske mulighetene er i de fleste tilfellene brukt i første omgang. Der hvor strålebehandling ikke er gjennomført, må det nå vurderes. Reoperasjon kan være aktuelt. Førstevalget i Norge for barn med residiv av medulloblastom, er Memmat-protokollen. Denne protokollen er nå åpen for inklusjon av norske pasienter. Dette er en metronomisk og målrettet anti-angiogenetisk behandlingsprotokoll, som også inkluderer intratekal cellegift.
Supratentoriell PNET
Andre embryonale hjernesvulster
Disse svulstene gikk tidligere under fellesbetegnelsen supratentoriell PNET (primitiv nevroektodermal tumor). Medulloblastom er en primitiv nevroektodermal tumor lokalisert i lillehjernen, mens disse andre ble kalt supratentoriell PNET da de var lokalisert supratentorielt. PNET er imidlertid ikke lenger en egen gruppe svulster i WHO-klassifiseringen fra 2016, men er nå delt opp i flere ulike entiteter i gruppen embyonale svulster (Louis et al., 2016).
Uspesifikke symptomer som tegn på økt intrakranielt trykk, kramper og nevrologiske utfall er det som oftest bringer pasienten til kontakt med helsevesenet.
Diagnosen stilles ved MR-undersøkelse. Hele CNS-aksen må visualiseres og vevsprøve fra svulsten samt cytologi fra spinalvæsken må undersøkes for endelig diagnose og klassifisering.
WHOs klassifikasjon av embryonale svulster som ikke er medulloblastomee (2016) inneholder følgende undergrupper:
- Embryonal tumor med rosetter, C19MC-endret (ETMR)
- Embryonal tumor med rosetter, NOS (ETMR)
- Medulloepiteliom
- CNS nevroblastom
- CNS ganglionevroblastom
- CNS embryonal svulst, NOS
- Atypisk teratoid/rhabdoid tumor (se eget kapittel)
- CNS embryonal tumor med rhabdoide trekk
Molekylærbiologiske markører
Tumormateriale vil bli undersøkt med tanke på mange molekykærbiologiske markører, noe avhengig av hvilken type embryonal svulst man mistenker. For eksempel vil CNS nevroblastom oftest ha gevinst av kromosomarm 1p, tap av kromosomarm 16q og FOXR2-forandringer,
Behandling
Behandling av disse embryonale svulstene krever høyspesialiserte og bredt sammensatte nevroonkologiske team (MDT). Behandlingen gjennomføres i henhold til internasjonale studieprotokoller/retningslinjer (B. H. Cohen et al., 1995; Massimino et al., 2006; Timmermann et al., 2006).
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Operasjoner bør derfor gjøres der det finnes erfarne nevrokirurger med omfattende erfaring med barn og god barneanestesi-/intensivekspertise.
Stråleterapi er en viktig del av behandlingen og for å ha et realistisk håp om kurasjon må hele nevralaksen bestråles med boost mot primærtumorområdet og eventuelle metastaser. Dose til nevralaksen vil for de fleste være ca 36 Gy, mens man til primærtumorområdet og eventuelle metastaser totalt gir 54 Gy (ca 50 Gy ved bestråling spinalt). Grunnet risiko for alvorlige senskader anbefales ikke bestråling til de minste barna (Timmermann et al., 2006). Mer spesifikt gir man for norske barn under 4 år ikke strålebehandling til nevralaksen, mens man vurderer om det er aktuelt med lokal bestråling mot tumortomt og eventuell resttumor for de som er fra 2 år og oppover. Ett av ankepunktene mot sistnevnte strategi er at det ved recidiv blir vanskeligere å gi ny strålebehandling. Stråling med protoner bør alltid vurderes.
Cytostatikabehandling er under kontinuerlig evaluering. Pasientene behandles oftest i henhold til HIT MED Guidance. Hos barn < 4 år startes det med postoperativ cytostatikabehandling i håp om å utsette stråleterapi lengst mulig.
Prognose
Prognosen varierer avhengig med type embryonal svulst og er vesentlig dårligere enn for medulloblastom. For hele gruppen (som tidligere ble benevnt PNET) ligger langtidsoverlevelsen ligger på ca 30–40 % (B. H. Cohen et al., 1995).
Oppfølging
Alle barn kontrolleres for tumorstatus etter 3, 6, 12 18 og 24 mnd med MR, siden MR hvert år til 5 år etter avsluttet behandling. Etter 5 år er det individuell oppfølging, men pga at de fleste har fått strålebehandling mot total CNS-akse, vil vi anbefale MR-kontroller livslangt.
I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. I varierende grad gjøres nevropsykologisk og spesialpedagogisk testing, undersøkelse av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering. Alle barn med PNET vil i varierende grad får senfølger av behandlingen som må følges nøye, og spesielt må endokrinologisk substitusjonsbehandling og pedagogisk tilrettelegging etableres.
Residivbehandling
Residivbehandling er nedslående. Der hvor strålebehandling tidligere ikke er gjennomført, må det nå vurderes. Reoperasjon kan være aktuelt. Utprøvende behandling med cytostatika kan startes, men resultatene så langt er ikke oppløftende.
Atypisk teratoid-rhabdoid tumor/ATRT
Atypisk teratoid / rhabdoid tumor beliggende i CNS (CNS AT/RT) er en svært ondartet svulst som hovedsakelig forekommer hos barn under tre år.
Diagnosen stilles ved histologisk undersøkelse.
Billedundersøkelse: Hele CNS-aksen må visualiseres og spinalvæsken undersøkes for klassifisering og utbredelse. Billedundersøkelse viser ofte cyster eller blødninger. På T1-vektet MR er svulstene hypointense, mens T2-vektede bilder viser iso- til hyperintense-lesjoner. Det er heterogen kontrastladning og ved leptomeningeal sykdom ser man ved billedundersøkelse en nodulær, klumpete kontrastoppladning langs meningene i spinalkanalen og inn i cauda equina. Billedundersøkelse alene kan ikke sikkert skille mellom AT/RT og suptratentorielle nevroblastomer (tidligere PNET) eller medulloblastom. Omtrent to tredjedeler av AT/RT-er forekommer i lillehjernen, vanligvis i den cerebellopontinvinkel, med infiltrasjon av omgivende strukturer. Omtrent en fjerdedel er supratentorial og 8 prosent er multifokale.
Patologi: Histologisk er AT/RT preget av rhabdoide celler og ligner på andre små runde blåcelletumorer. AT/RT kan imitere flere forskjellige entiteter; også medulloblastomer Nekrose og en høy grad av mitotisk aktivitet er vanlig. Germcellemarkører er negative.
Biologiske markører: Diagnostisering av AT/RT krever tap av enten INI1 kjernefarging, noe som indikerer biallel inaktivering av SMARCB1 på kromosom 22, eller tap av BRG1-farging, noe som indikerer SMARCA4 inaktivering. I tillegg til disse somatiske mutasjonene påvist i tumorvev, har omtrent en tredjedel av pasientene kimcelle mutasjoner i SMARCB1 eller, mindre vanlig, SMARCA4. Vanligvis er de minst delvis vimentin, CK, EMA og smooth muscle actin (SMA) positive. Ki67 er vanligvis relativt høy. Med hjelp av OTX2 fargning er det mulig å undersøke om det dreier om en AT/RT-Tyr. /flere av disse markørene vil kunne bli implementert som prognostiske faktorer i fremtidig protokoller (Japp, Klein-Hitpass, Denkhaus, & Pietsch, 2017; Johann et al., 2016).
Epidemiologi
AT/RT er den mest vanlige ondartede CNS tumores hos barn under 1 år, og AT/RT representerer omtrent 1–2 % hjernesvulster hos barn. I Norge har det vært 19 tilfeller av AT/RT i de 15 årene 2002–2017, det tilsier i snitt drøyt en pasient i året i hele Norge.
Symptomer: Symptomer hos spedbarn er ofte slapphet, oppkast og eller økende hodeomkrets; eldre barn kan ha symptomer grunnet affeksjon av hjernenervene IV, VI eller VII, for eksempel torticollis, dobbeltsyn eller facialisparese. Hemiplegi og/eller hodepine generelle trykksymptomer kan også være debutsymptomer.
Prognosen: I de fleste studier og rapporter er dårlig, med en median overlevelse på 12 til 14 måneder og fem års samlet overlevelse mindre enn 50 prosent. Pasienter med familiemutasjoner i SMARCB1genet har en tendens til å være yngre og ha dårligere prognose sammenlignet med de med ikke familiære, sporadiske svulster. Faktorer som er assosiert med bedre prognose inkluderer supratentoriell beliggenhet, ikke spredning av sykdommen ved diagnosetidspunktet, fullstendig reseksjon av tumor kirurgisk og uttrykk for ASCL1, en regulator for NOTCH-signalering.
Behandling: Kirurgi alene er ikke kurativt ved AT/RT, men en kombinasjon av kirurgi, omfattende cellegiftbehandling og strålebehandling er helt nødvendig for overlevelse (Fossey et al., 2017; Richardson, Ho, & Huang, 2018; Schrey et al., 2016).
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Målsetningen med kirurgi ved AT/RT som ved andre ondartede CNS svulster er maksimal reseksjon.
Cytostatikabehandling er under kontinuerlig evaluering, og til nå har vi fulgt en anbefaling gitt i «A multinational registry for rhabdoid tumors of any anatomical site EUROPEAN RHABDOID REGISTRY, EU-RHA». AT/RT registeret er en anbefaling både når det gjelder utredning, behandling og oppfølgning av AT/RT. Her anbefales det å gi 9 kurer med kjemoterapi hver 2. uke, dels intravenøst og dels også intrathekalt i Ommaya reservoir. AT/RT svulstene er vanligvis initialt kjemosensitive. Alle barn får cytostatika under strålebehandling, men kurene modifiseres under strålebehandlingen på grunn av fare for økt toksisitet. I anbefalingene som er gitt i dette registret, har man også foreslått høydosebehandling med stamcellestøtte. Til nå har ikke høydosebehandling vist noen signifikant bedret overlevelse enn de 9 regulære kjemoterapikurer. Etter strålebehandling skal man ikke lenger gi intrathekal kjemoterapi.
Stråleterapi er en viktig del av behandlingen for å ha et realistisk håp om kurasjon, og man anbefaler å komme i gang med strålebehandlingen så tidlig som mulig i forløpet, i løpet av de første kurene. Ved ung alder og ikke dissiminert sykdom, anbefales lokal stråling mot tumorsengen, men ved eldre alder, resttumor og disseminert sykdom, anbefales å stråle hele nevralaksen med boost mot primærtumorområdet og eventuelle metastaser. Grunnet risiko for alvorlige senskader anbefales ikke CNS akse bestråling til de minste barna. Man anbefaler stråling med protoner.
Videre studier/planlagt behandling: Det er en planlagt protokoll med stratifiseringer og randomiseringer på trappene, men den kommende AT/RT protokoll er ikke offisiell ennå. Den vesentlige endringen er å gi 12 runder kjemoterpi i stedet for 9, : 3 dox, 4 VCA og 5 ICE.
Oppfølging: Alle barn kontrolleres med tanke på tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, deretter MR minimum to ganger årlig fra 2 til 5 år etter avsluttet behandling, og årlig til 10 år etter ferdig behandling. De barna som ikke har fått strålebehandling, anbefales å ta spinalpunksjon 2 ganger i året i 2 år etter ferdig behandling.
I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. Alle barn med AT/RT vil i varierende grad få senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprofgram for barn med tumor cerebri og det gjøres en vurdering av behov også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. I varierende grad får barnet oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering.
Residivbehandling: Ved residiv er behandlingsresultatene nedslående. De terapeutiske mulighetene er i de fleste tilfellene brukt i første omgang. Der hvor strålebehandling ikke er gjennomført, må det nå vurderes. Reoperasjon er sjelden aktuelt, da residivet ofte viser rask vekst, og tumor ikke lar seg fjerne. MEMMAT protokoll med metronomisk behandling med blant annet Avastin, peroral kjemoterapi, intrathekal behandling og diverse perorale medisiner (Celecoxib, Fenofibrat, Taidomid) kan vurderes. Målrettede terapier er også under utredning i en rekke fase-1 studier, og et stort antall medikamenter har potensiell terapeutisk aktivitet. Spesielt Aurora kinase A-hemmeren Alisertib, (MLN8237) har vist antitumoraktivitet hos barn med tilbakefall av AT/RT eller resistent sykdom.
Germinalcellesvulster i CNS
Germinalcelletumores (GCT) er en gruppe sjeldne svulster med antatt utgangspunkt i totipotente primordiale germinalceller, som migrerer til «feil plass» og kan undergå malign transformasjon i løpet av vekst og differensiering. De er heterogene både mht primærlokalisasjon, histologi, biologisk profil og behandlingsrespons. Totalt utgjør GCT ca. 3,4 % av maligne svulster hos barn, og ca. 1/5 av dem oppstår i sentralnervesystemet. Insidenskurven er to‑puklet, høyest hos sped-/småbarn med en ny stigning rundt puberteten, og de er hyppigere hos gutter
Lokalisasjon
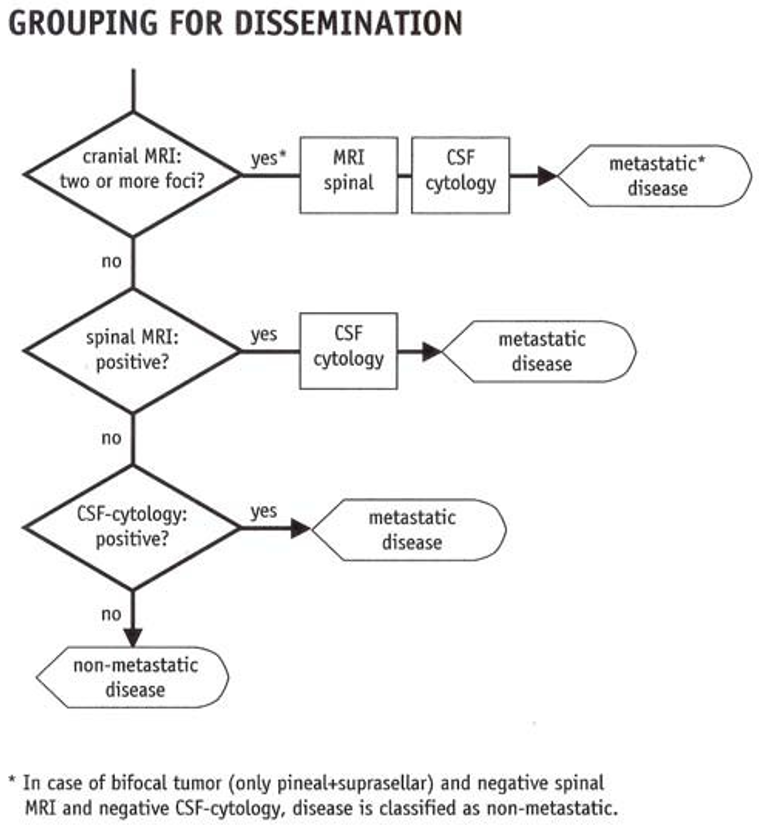
De finnes oftest i midtlinjen, selv om andre lokalisasjoner forekommer. Særlig vanlig er pinealregionen. Undergruppen «germinomer» kan være bifokale (pinealregion + suprasellært) uten at dette regnes som metastatisk sykdom. Vanligste spredningsform er via utsæd i cerebrospinalvæsken.
Symptomer/tegn
Avhenger av lokalisasjon. Ved beliggenhet i pinealregionen oppstår ofte hydrocephalus med trykksymptomer pga aqueductstenose. Videre sees ofte øyepareser og diplopi, samt «Parinauds syndrom» (blikkparese oppad og dissossiert pupillerefleks med intakt akkomodasjon, men manglende lysrefleks). Ved suprasellær lokalisasjon omfatter symptombildet foruten synsforstyrrelser også ofte diabetes insipidus og andre endokrine dysfunksjoner, som f.eks. forsinket pubertet.
Diagnose
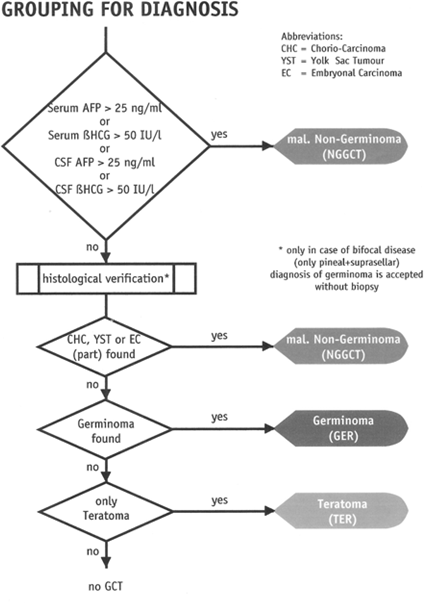
Ved mistanke om intracerebral GCT bør pasienten utredes videre ved spesialavdeling. MR-undersøkelsen må omfatte hele CNS-aksen i henhold til spesifiserte retningslinjer i SIOP CNS GCT II-protokollen. Spinalvæsken skal undersøkes cytologisk for utsæd av tumorceller. Det må gjøres før tumor er manipulert operativt. Dersom det har vært behov for ø.hj.-operasjon pga høyt intracranielt trykk, bør det gå minst 10 dager før undersøkelsen foretas. Tumormarkørene AFP og Beta-HCG skal analyseres i både serum og cerebrospinalvæske. Dersom disse er forhøyet (i én eller begge komponenter), kan man fastslå at det dreier seg om en «secreting germ cell tumor» i undergruppen malignt non-germinom (NG-GCT). Dermed kan biopsi unngås. Men dersom markørene er under de definerte grenseverdiene kreves histologisk verifikasjon, dog med unntak av bifocal sykdom med karakteristisk lokalisasjon, som er typisk for germinom (se ovenfor).
Utredningen må omfatte så vel utbredelsesgrad som histologisk subtype, som skissert i følgende figurerer fra SIOP-CNS GCT II-protokollen.
Rekkefølgen av undersøkelsene avhenger av om det foreligger behandlingstrengende akutte symptomer:
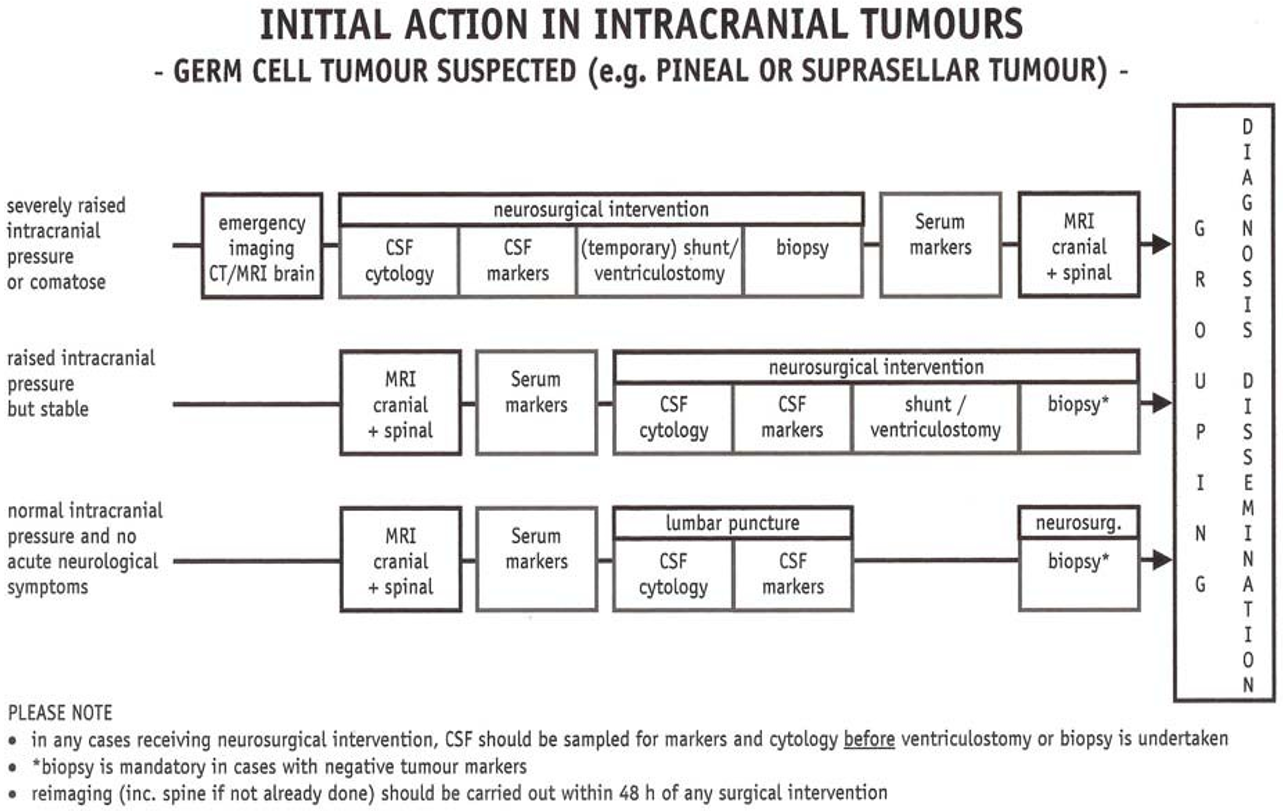
Patologi/Histologi
Germinalcellesvulster kan fremvise:
- Germinalcelle differensiering (→germinom, som tilsvarer seminom og dysgerminom i testis)
- Somatisk diffensiering:
- Embryonal differensiering (→ embryonalt carcinom)
- Ekstraembryonal (→choriocarcinom og → plommesekktumor = yolksac tumor = YST)
- Teratom (moden/umoden) med tilstedeværelse av modne eller umodne strukturer fra alle 3 germinal-lag (graderes 1–3 avhengig av relativ mengde umodent vev).
NB: Ofte forekommer blandingsformer!
Histologien hos nyfødte er nesten alltid teratom (modne og umodne), i blant med små foci av plommesekktumor. I spedbarns- og småbarnsalder dominerer YST, mens andel germinomer stiger i senere barne- og ungdomsår.
Tumormarkørene har tendens til overlapping mellom undergruppene, teratomceller kan f.eks. være noe positive for AFP. Det er avgjørende viktig for valg av behandling å undersøke for et bredt immunhistokjemisk/molekylærbiologisk/cytogenetisk panel.
Histologi | Klinikk | Tumormarkører | Følsom for | ||
|---|---|---|---|---|---|
|
| AFP | Tot. HCG | Cytostatika | RT |
Germinom | malign | – | (+) | +++ | +++ |
Malignt non-germinom (secreting GCT): |
|
|
|
|
|
Embryonalt carcinom | malign | – | – | +++ | ++ |
Plommesekktumor | malign | +++ | – | +++ | ++ |
Choriocarcinom | malign | – | +++ | +++ | ++ |
Teratom | benign (potensielt malign) | – | – | –/? | +/– |
Behandling
Både utredning og behandling av GCTs krever nevroonkologiske spesialistteam, og pasienten må snarest mulig henvises til et universitetssykehus.
Prinsippet for valg av terapi ved sammensatte GCTs er at man skal behandle i henhold til den mest maligne komponenten. Dersom full utredning mht markører og utsæd ikke er foretatt, må sykdommen behandles som metastatisk sykdom av den mest maligne undergruppe.
Det er meget viktig å sikre en fullverdig utredning nært opptil oppstart av behandling (min. 14 dager før, ellers kreves ny utredning).
Protokollen SIOP CNS GCT II er stengt for inkludering av nye pasienter, men brukes fortsatt som utgangspunkt for behandlingen av maligne germinalcellesvulster. En ny protokoll er under utarbeidelse, men avhenger av resultatene av nåværende protokoll (analyseres etter follow up avsluttes i 2020).
Generelt avhenger behandlingsintensiviteten av histologi, utbredelsesgrad, AFP-nivå og alder (grense 6 år). Strategien må korrigeres avhengig av evalueringer underveis, bl.a. med henblikk på behandlingsrespons.
Nevrokirurgi er viktig for biopsi der dette er påkrevet, samt ved behov for trykkavløsende ventriculostomi. Ved rene teratomer er kirurgi hovedbehandlingen. Kirurgisk reseksjon kan også være aktuelt ved resttumor etter kjemoterapi for andre histologiske undergrupper.
Teratom behandles i hovedsak med kirurgi. Evt. tilleggsbehandling individualiseres etter histologi, reseksjonsstatus mv. Studiesentret kan kontaktes for råd.
Lokalisert germinom behandles med kjemoterapi og ventrikulær strålebehandling med fokal boost, uansett om de er i komplett eller partiell remisjon etter kjemoterapi (ihht partiell remisjon etter kjemoterapi i protokollen (standard behandling)).
Germinom med metastaser behandles med strålebehandling mot total-CNS (CSI) ihht SIOP CNS GCT II-protokollen.
Lokalisert NG-GCT behandles med kjemoterapi og strålebehandling. Kjemoterapien er PEI-kurer (Cisplatin, Etoposid og Ifosfamid) ihht standard risiko pasienter i protokollen. For høy-risiko NG-GCT (AFP>1000 ng/ml el. diagnosealder < 6år) diskuteres valg av kjemoterapi. Studiesentret kan kontaktes for råd. Strålebehandlingen er lokal ihht SIOP CNS GCT II-protokollen, men inkludering av ventriklene vurderes, spesielt gjelder dette ved protonbestråling.
NG-GCT med metastaser behandles med kjemoterapi og strålebehandling. Kjemoterapien er PEI-kurer (Cisplatin, Etoposid og Ifosfamid) ihht standard risiko pasienter i protokollen. For høy-risiko NG-GCT (AFP>1000 ng/ml el. diagnosealder < 6år) diskuteres valg av kjemoterapi. Studiesentret kan kontaktes for råd. Strålebehandlingen mot total-CNS (CSI) ihht SIOP CNS GCT II-protokollen.
Prognose
Best prognose har germinom, med 90–95 % 5-års EFS i foregående SIOP-protokoll (CNS GCT-96) som inneværende bygger videre på. Ved NG-GCT er EFS rundt 70 %.
Oppfølging
Detaljert oppfølgningsplan både m.h.t residiv og senskader foreligger i protokollen. Residiv ved maligne GCTs er hyppigst de første 2 år etter behandling. Alle pasienter skal følges opp (inkl. klinikk, MR og markører) hver 3. måned første år og videre i h.h.t. protokoll minst 5 år etter avsluttet behandling. Ved germinom (som kan residivere enda senere) anbefales oppfølgning minst 10 år.
Barn med germinalcelletumor vil i varierende grad få senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri, og det gjøres også en vurdering av behov for dette tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. I varierende grad får barnet oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering.
For mer informasjon, se referanselisten til «SIOP CNS GCT II protocol for the diagnosis and treatment of children, adolescents and young adults with intracranial Germ Cell Tumours» (Prospective Trial for the Diagnosis and Treatment of Intracranial Germ Cell Tumors (SIOPCNSGCTII), 2011-), hvor erfaringer fra en lang rekke europeiske, amerikanske og japanske studier er omtalt (Fullstendig protokoll tilgjengelig på hjemmesidene til KSSB).
Kraniofaryngeom
Kraniofaryngeom er en histologisk benign epitelial svulst utgående fra embryonale rester av ductus craniopharyngeus.
På tross av benign histologi fører slike svulster hos mange pasienter til betydelige senskader i form av hypopituitarisme, massiv overvekt, synsforstyrrelser og kognitive skader. Dette skyldes beliggenheten i hypothalamus og nærhet til synsveiene.
Kraniofaryngeomer klassifiseres til WHO grad I.
Etiologien er ukjent og det foreligger ingen kjente genetiske disposisjoner.
Forekomst
Kraniofaryngeomene utgjør ca. 3 % av intrakranielle svulster hos barn, dvs 1–2 nye tilfeller i Norge pr år. Ingen kjønnsforskjell.
Det er to ulike manifestasjoner ved vevsundersøkelse, adamantinomatøs type som er helt dominerende hos barn, mens voksne oftest har papillær type.
Molekylærbiologi
Det er nylig påvist at barnekraniofaryngeom, den adamantinomatøse typen, har en mutasjon i genet til beta-catenin som er viktig for wnt-signalveien. Mutasjonen fører til overaktivering av beta-catenin og derved wnt. Dette aktiverer også andre signalveier «downstream» i påvirkningsrekken med tumorutvikling.
Lokalisasjon
Vanligst suprasellært, evt med intrasellær komponent. Kan være rent intrasellær, mens enkelte har svær, gjerne cystisk utbredelse, også til 3. ventrikkel. Ektopisk lokalisasjon (ekstradural) forekommer.
Symptomer
Synsaffeksjon som uttrykk for kompresjon på synsbanekrysningen og synsbaner. Vekst i hypothalamus og hypofyse gir hormonelle forstyrrelser, herunder ikke sjelden vekstforstyrrelser.
Noen debuterer med diabetes insipidus. Hypothalamuslesjon kan gi kraftig overvekt og også kognitiv svikt. Ved store svulster økt intracranielt trykk evt med hydrocephalus.
Diagnose og utredning
MR undersøkelser gir vanligvis avklarende diagnostikk. CT kan vise kalk. Ved stor cyste tappes vanligvis fra denne med funn av «maskinoljelignende» væske. Biopsi gir sikker diagnose.
Hormonutredning av hypothalamus-hypofyseaksen og diabetes insipidus er viktig. Likeledes synsundersøking, vekstforhold og kognitiv status.
Behandling
Internasjonalt har man ikke kommet frem til en klar og entydig algoritme for behandling av denne vanskelig tumortypen (Muller, 2010). Hvert enkelt barn må vurderes for et individualisert behandlingstilbud basert på vurderinger ut fra pasientens alder, kliniske tilstand og tumors størrelse og vekstmønster.
Kirurgi har tradisjonelt vært primær behandling hvor man har tilstrebet radikalitet. Det er imidlertid en økende internasjonal trend å være skånsom ved reseksjon for å unngå skade på hypothalamus (Puget et al., 2007). Hypothalamuslesjon er en følge av tumors infiltrasjon, men det har vist seg at ved kirurgi i denne regionen kan sekvelene øke betydelig. En tilstreber derfor kirurgi av tumordelen utenfor hypothalamus, ikke minst hvis det er påvirkning av synsveiene. Biopsi gjøres ved diagnose.
Ved inkomplett reseksjon kan postoperativ strålebehandling / stråleknivbehandling være aktuelt for å redusere risiko for videre vekst. En større tysk studie viser at en ikke taper noe ved å utsette en slik behandling inntil det har inntruffet vekst av tumor etter kirurgien.
Intracystisk behandling. Hos barn er de fleste svulstene cystiske og ved store cyster kan man initialt implantere kateter for tømming av cystene og avlasting av press på omgivelsene. Instillasjon i cystene av cellegift (bleomycin) eller isotop (Yttrium) har tidligere vært brukt i behandlingen. Komplikasjonene har imidlertid vært utfordrende og behandlingen komplisert. I de senere år har instillasjon av interferon blitt standardbehandling ved flere sentra. Man oppnår ikke radikal fjerning av tumor, men avlastning av synsnerver og omkringliggende vev med stor grad av symptomfrihet. Pasienten må følges med regelmessige billedoppdateringer til nydanning av cystene opphører og hvis det ikke er mulig å kontrollere cystene med tapping/ interferon kan det være indisert med annen kirurgisk behandling evt i kombinasjon med stråleterapi.
Systemisk medikamentell behandling er i dag på forsøksstadiet. Signalveier «downstream» for wnt-signalveien aktiveres ved kraniofaryngeom, for eksempel MAPK-signalveien (RAS/RAF/MEK/ERK). En har derfor nå i England startet forsøk med MEK-inhibitor i utvalgte tilfeller. Resultater foreligger ikke nå.
En annen tilnærming er antiinflammatorisk behandling, da inflammasjon er en viktig del av patogenesen ved disse svulstene. Aktuelt er Interleukin-1 Reseptor-inhibitor (anakinra). Kliniske forsøk vil komme.
Symptomatisk behandling: Det er vesentlig at barn med slike svulster følges opp i et multidisiplinært team (MDT) med bred erfaring innen nevrokirurgi, nevroonkologi og nevroendokrinologi. Hormonutfall må monitoreres og substitueres. Vektøkning må prøves begrenset Videre er det viktig med oppfølging av kognitive funksjoner, synsfunksjon osv.
Prognose
Selv etter radiologisk tilsynelatende radikal kirurgisk reseksjon ser man opp til 15 % residiv. Pasientene som er behandlet med intracystisk interferon har svært gode resultater hva angår symptomkontroll, men oppfølging blir som ved en kronisk sykdom. Mortaliteten ved kraniofaryngeom er svært lav, men som fremgår er det høy morbiditet.
Oppfølging
Oppfølging avhenger av valgt behandlingsregime. Ved både cystebehandling og tradisjonell kirurgisk behandling er det tett oppfølgning, da residiv forkommer hyppig de første 5 år. Det er risiko for sent residiv, så kontroller (klinisk og MR), bør gjennomføres med lengre intervall gjennom flere tiår.
Nøye symptomatisk oppfølging er helt vesentlig.
Barn/ungdom med kraniofaryngeom kan i varierende grad få senfølger av behandlingen som må følges. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprofgram for barn med tumor cerebri, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering når det er behov for dette.
Plexus chorioideus-svulster
Epidemiologi
Plexus choroideus svulster utgjør kun 2–4 % av hjernesvulstene hos barn. De fleste pasientene er mellom 0–3 år på diagnosetidspunktet.
Symptomer
Små barn har som oftest en raskt økende hodeomkretsutvikling, eldre barn utvikler generelle trykksymptomer.
Billedundersøkelse
Svulsten er utgående fra plexus choroideus, vevet som produserer CSF i hjernes naturlige hulrom. Svulster i plexus choroideus gir overproduksjon av CSF og ledsagende hydrocephalus er et typisk funn. Hos små barn ser man svulsten i en av sideventriklene eller tredje ventrikkel. Hele CNS-aksen må avbildes for eventuelle metastaser (carcinomer).
Patologi/ Klassifisering
WHO klassifiseringen skiller mellom 3 ulike histologiske grader: Plexus choroideus papillom (WHO grad I), atypisk plexus choroideus papillom (WHO grad II) og plexus choroideus carcinom (WHO grad III).
Diagnosen stilles ved histologisk undersøkelse.
Prognosen
Plexus choroideus papillomer har ved total kirurgisk fjerning en svært god prognose (kurativt).
Plexus choroideus carcinomer har i de fleste rapporter en 5 års overlevelse på under 50 %.
Behandling
Kirurgi er grunnlaget for behandling av plexussvulster hvor målet er total fjerning av tumor. De fleste pasientene er på behandlingstidspunktet i første leveår og har et lite blodvolum, svulstene har stor blodgjennomstrømning, som oftest med en betydelig hydrocefalus. Det kirurgiske inngrepet er derfor utfordrende. Mange vil kreve oppfølgning og eventuell behandling for hydrocephalus i forløpet.
Pasienter med plexus papillom (WHO grad I og II) skal ikke ha tilleggsbehandling, men skal følges opp over tid (regelmessige MR undersøkelser)
Plexus choroideus carcinom (WHO grad III) er en aggressiv tumor. Prognosen er noe bedre ved total fjerning av tumor. Mange har sykdom med spredning og eller innvekst sentralt i hjernen allerede på diagnosetidspunktet og vil være avhengig av tilleggsbehandling.
Plexus choroideus carcinomene er følsomme for kjemoterapi med kombinasjoner av carboplatin, etoposid, vincristin og cyklofosfamid. Stråleterapi forbedrer prognosen, men pasientens alder vil ofte være en begrensende faktor (de fleste vil være for unge). Det er ingen konsensus omkring optimal behandling.
Oppfølging: Alle barn kontrolleres med tanke på tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, deretter MR minimum to ganger årlig fra 2 til 5 år etter avsluttet behandling, og senere årlig.
Barn med plexuscarcinomer vil i varierende grad utvikle senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres i hht nasjonalt tverrfaglig oppfølgingsprogram for barn med hjernesvulst 1, 2 og 5 år etter diagnose, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig.
Det anbefales oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering.