Sarkomer
Sarkomer er maligne svulster som utgår fra mesenkymalt vev. Det er den nest vanligste formen for solid, malign svulst utenfor sentralnervesystemet i barnealder (Childhood cancer in the Nordic countries, 2008).
Sarkomer inndeles i bensarkomer og bløtvevsarkomer og får navn etter det vevet de har fellestrekk med.
Bensarkomer inndeles i osteogent sarkom (Kager et al., 2003) og Ewing sarkom, sistnevnte kan også opptre i bløtvev (Principles and practice of pediatric oncology, 2006).
Bløtvevsarkomer inndeles i rabdomyosarkom som utgår fra celler som er eller skal bli muskelceller, og non-rabdomyosarkom. Non-rabdomyosarkom er en heterogen gruppe tumores som består av mer enn 30 ulike undergrupper (Principles and practice of pediatric oncology, 2006).
Se også «Nasjonalt handlingsprogram med retningslinjer for utredning, behandling og oppfølging av sarkom» (Helsedirektoratet, 2018).
Epidemiologi
Insidensen av sarkom hos barn er 1,49 pr 100 000, det vil si at 9,5 % av barna som får en kreftdiagnose, har sarkom. Bløtvevsarkom er hyppigst og forekommer hos 6 % av de kreftsyke barna. Bensarkom er sjeldnere og utgjør 3,5 % av krefttilfellene hos barn under 15 år (Childhood cancer in the Nordic countries, 2008).
Symptomer og funn
Osteogent sarkom debuterer oftest med smerter. I begynnelsen er smertene ofte intermitterende, men etter hvert blir de konstante og mer intense. Tumorrelatert hevelse og nedsatt funksjon i naboledd oppstår mye senere. I ca 10 % av tilfellene debuterer osteogent sarkom som patologisk fraktur (Kager et al., 2003).
Ewing sarkom debuterer oftest med smerter. Smertene er vedvarende og kan oppstå uavhengig av aktivitet, for eksempel i hvile eller om natten. Eventuelle tilleggssymptomer avhenger av tumors lokalisasjon og størrelse. Subfebrilitet er rapportert hos ca 1/3 av pasientene, først og fremst hos de med metastatisk sykdom på diagnosetidspunktet (Paulussen, Kovar, & Jürgens, 2005).
Hvordan et bløtvevsarkom debuterer, avhenger av tumors lokalisasjon og størrelse. Hvis ikke andre strukturer komprimeres, gir sarkom sjelden uttalte symptomer og oppdages ofte tilfeldig grunnet tumors størrelse.
Bløtvevsarkom kan gi smerter hvis de presser på andre strukturer eller obstruerer avløp som for eksempel i blæreregionen. Vedvarende nesetetthet og sekresjon fra nesen er vanlige symptomer ved sarkom i nasofarynks. Vaginale polypper og utflod er vanlig ved sarkom i uterus og vagina (Bisogno & Bergeron, 2005).
Allmenn sykdomsfølelse og vekttap forekommer svært sjelden i startfasen ved sarkom, men forekommer hyppigere ved avansert sykdom.
Utredning
Utredning av sarkom hos barn og ungdom bør skje ved Barne-/ungdomsavdelinger med spesialkompetanse i håndtering av sarkom eller et sarkomsenter. Det er svært viktig at utredningen skjer i henhold til gjeldende protokoller. Man bør mistenke sarkom ved tumor i benvev eller tumor beliggende dypt i bløtvev.
Utredning med tanke på diagnostikk og stadieinndeling gjøres i henhold til aktuelle behandlingsprotokoller og må inneholde adekvat billeddiagnostikk av primærtumor og nøyaktig evaluering av potensielle metastaser.
Radiologiske undersøkelser
Ved mistanke om osteogent sarkom er det viktig med vanlig røntgenundersøkelse av det aktuelle området, gjerne supplert med CT for å se på skjelettstruktur, og MR for å evaluere benmargs- og bløtdelsaffeksjon samt tumors relasjon til årer og nerver. Søk etter metastaser konsentreres om skjelett og lunger for >95 % av metastasene ved osteogent sarkom opptrer i disse lokalisasjonene. Røntgen og CT av lungene benyttes for å kartlegge eventuelle lungemetastaser. MR av affisert knokkel skal utføres for å lete etter skip-metastaser. MR og NaF-PET bør utføres for å kartlegge eventuelle skjelettmetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved osteogent sarkom.
Ved utredning av Ewing sarkom benyttes vanlig røntgen, MR og CT for å kartlegge sykdoms-utbredelse på primærtumorstedet. CT egner seg best til å vise endringer i korteks- og skjelettstruktur, mens MR egner seg best for kartlegging av tumorutbredelse i benmarg og bløtvev samt for å evaluere tumors relasjon til nærliggende strukturer som årer og nerver. Det er viktig å dokumentere tumors utbredelse i 3 dimensjoner. CT thorax anbefales for kartlegging av eventuelle lungemetastaser. MR anbefales for kartlegging av eventuelle skjelettmetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved Ewing sarkom.
Ved mistanke om bløtvevsarkom bør man benytte MR, ultralyd og eventuelt CT av primærtumor-stedet. Det er viktig å kunne angi tumors størrelse i 3 dimensjoner ved diagnose, slik at man kan evaluere behandlingsrespons ved senere evalueringstidspunkter. MR er å foretrekke for de fleste lokalisasjoner og er obligatorisk ved primærtumor i genitourinaltraktus og ved paraspinal tumor. CT kan være et supplement ved spørsmål om subtile bendestruksjoner. Røntgen thorax og CT thorax benyttes for å oppdage lungemetastaser. Ultralyd abdomen benyttes for å kartlegge lymfeglandelaffeksjon og levermetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved bløtvevsarkom.
Biopsi
Sarkomdiagnosen må alltid bekreftes histologisk. Målet med biopsitaking er å få nok tumormateriale til histologi, immunhistokjemi, cytogenetikk, sentralisert «review», samt vev i reserve for eventuelle supplerende analyser.
Biopsitaking skjer oftest ultralydveiledet, men kan også skje CT-veiledet. Det er nyttig å starte med finnålsaspirasjon og fortsette med tru-cut-biopsi hvis forholdene ligger til rette for det. Dersom denne fremgangsmåten ikke kan sikre nok materiale for å kunne stille en sikker diagnose er det aktuelt å supplere med åpen biopsi. Det er viktig å ta med i betraktning at biopsikanalen og eventuelle arr må inkluderes «en-bloc» ved etterfølgende kirurgi, så det er viktig at affeksjon av nevrovaskulære strukturer i størst mulig grad unngås. På ekstremiteter bør incisjoner legges longitudinelt. Farging f.eks. med metylenblått bør benyttes for markering av biopsikanal. Endoskopisk biopsitagning er akseptabelt ved tumor i blære, prostata og vagina.
Benmargsaspirasjon og eventuelt benmargsbiopsi hører med i utredningen av enkelte typer bløtvevsarkom.
Spinalpunksjon for å sjekke om det foreligger CNS-affeksjon, er kun nødvendig ved CNS-nær tumor.
Behandling
Sarkom i barnealder utredes og behandles i henhold til internasjonale protokoller. For øyeblikket behandles osteogent sarkom etter anbefalinger fra den avsluttede EURAMOS 1-protokollen, Ewing sarkom behandles i henhold til EuroEwing 2012-protokollen, mens residiv av Ewing sarkom og refraktært Ewing sarkom behandles i henhold til rEECur-protokollen. Rabdomyosarkom behandles i henhold til anbefalinger fra den avsluttede EpSSG RMS 2005-protokollen og høygradig malignt non-rabdomyosarkom behandles i henhold til anbefalinger fra den avsluttede EpSSG NRSTS 2005 (https://www.skion.nl/). Når det gjelder rabdomyosarkom, har fagmiljøet i Norge søkt REK og SLV om å få godkjent at norske pasienter kan inkluderes i og behandles i henhold til den nye internasjonale behandlingsprotokollen fra EpSSG, nemlig «FaR-RMS: An overarching study for children and adults with Frontline and Relapsed RhabdoMyoSarcoma». Forhåpentligvis kan norske pasienter få tilbud om være med i denne studien allerede i løpet av 2020. Studien er i regi av EpSSG og behandlingen er basert på resultatene fra RMS-2005-protokollen. Behandlingen av sarkom i barnealder er i hovedtrekk multimodal og består av kjemoterapi, kirurgi og/eller bestråling.
Medikamentell behandling
Behandlingen av osteogent sarkom består av pre- og postoperativ kjemoterapi samt kirurgi. Kjemoterapien baserer seg på høye doser metotreksat, cisplatin og doksorubicin (Bielack et al., 2015; Smeland et al., 2019)
Ewing sarkom er generelt en kjemosensitiv tumorform og induksjonsbehandlingen består per i dag av vinkristin, doksorubicin, cyklofosfamid, ifosfamid og etoposid (EE2012-protokollen: https://birmingham.ac.uk/research/activity/mds/trials/crctu/trials/EE2012/index.aspx). Ved dårlig histologisk eller radiologisk respons, ved stor primærtumor (>200 ml) eller ved metastatisk sykdom kan man vurdere å intensivere kjemoterapien med høydosebehandling med autolog stamcellestøtte (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf). Ved behandlingsrefraktær sykdom og residiv består behandlingen av intensiv kjemoterapi og det er flere ulike kjemoterapiformer som har vist seg å ha effekt:
- cyklofosfamid/topotekan,
- irinotekan/temozolamid,
- gemcitabin/docetaxel og
- høydose ifosfamid.
I rEECur-studien randomiseres pasientene mellom de ulike behandlingsarmene for å finne ut hvilken av de fire armene som fungerer best. Foreløpige resultater fra rEECur-studien tyder på at gemcitabin/docetaxel har dårlig effekt og denne behandlingen anbefales derfor ikke.
Rabdomyosarkom er som regel en kjemosensitiv tumorform, og kjemoterapi benyttes derfor i behandlingen hvis man ikke kommer til målet med kirurgi alene. Kjemoterapi benyttes for å redusere tumorstørrelsen slik at radikal kirurgi, evt annen lokalbehandling, kan bli mulig. Aktuell kjemoterapi består av ifosfamid, vinkristin, actinomycin D og eventuelt doxorubicin. Etter avsluttet primærbehandling kan det være aktuelt med vedlikeholdsbehandling med cyklofosfamid og vinorelbin i lave doser (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf). I den nye behandlingsprotokollen (FaR-RMS) vil dessuten irinotekan, temodal og regorafenib inngå i behandlingen av enkelte pasientgrupper.
Non-rabdomyosarkom er en svært heterogen gruppe sarkomer, og de fleste er ikke like kjemo-sensitive som rabdomyosarkom. Den europeiske studien EpSSG NRSTS 2005 (skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf) inndeler non-rabdomyosarkom i 3 ulike hovedgrupper; synovialt sarkom, «voksen type» bløtvevsarkom og andre typer bløtvevsarkom. Synovialt sarkom er desidert den vanligste formen for non-rabdomyosarkom i barnealder. Denne sarkomtypen er relativt kjemosensitiv, og hvis det ikke ligger til rette for radikal kirurgi i utgangspunktet, anbefales det kjemoterapi med ifosfamid/doxorubicin for å redusere tumorstørrelsen før eventuell kirurgi. Ved «voksen type» bløtvevsarkom er effekten av kjemoterapi mer usikker, men EpSSG NRSTS 2005 anbefaler kjemoterapi i form av ifosfamid/doxorubicin dersom man ikke kan foreta radikal kirurgi i utgangspunktet (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Rabdoid tumor utenfor sentralnervesystemet er en undergruppe av non-rabdomyosarkom. Den er sjelden, men svært aggressiv og ofte letal i barnealder. Grunnet høy dødelighet av denne tumortypen er det svært viktig med adekvat behandling. EpSSG NRSTS 2005 anbefaler et intensivt kjemoterapiregime med vinkristin/doxorubicin/cyklofosfamid alternerende med ifosfamid/etoposid hver annen uke (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf). Alternativt kan rabdoid tumor behandles i henhold til anbefalingene fra det multinasjonale European rhabdoid registry (EU-RHAB). EU-RHAB anbefaler et intensivt kjemoterapiregime med doksorubicin, ifosfamid, karboplatin, etoposid, vinkristin, actinomycin D og cyklofosfamid, evt supplert med høydosebehandling med karboplatin og thiotepa, etterfulgt av autolog stamcellestøtte.
Kirurgisk behandling av rabdomyosarkom (RMS)
Lokal kontroll er nødvendig for å kurere barn med RMS, og dette kan oppnås ved kirurgi og/eller strålebehandling (evt brachyterapy). Konservativ tilnærming er anbefalt, og preoperativ kjemoterapi for reduksjon av tumor er vanlig før tumorreseksjon/bestråling.
Målet med lokalbehandling er kurasjon uten eller med minst mulig senskader. Hvilken type lokalbehandling som egner seg best avhenger av primærtumors lokalisasjon og størrelse, pasientens alder og kjemoterapirespons. Kirurgisk planlegging må inkludere alle rekonstruktive prosedyrer med optimal timing av eventuell strålebehandling i tillegg (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf).
Anbefalinger:
- Sarkom i barnealder utredes og behandles i henhold til internasjonale protokoller som krever samtykke fra foreldrene.
- Osteogent sarkom behandles i henhold til anbefalinger i den avsluttede EURAMOS 1-protokollen.
- Ewing sarkom behandles primært i henhold til EuroEwing 2012-protokollen og i henhold til rEECur-protokollen ved residiv.
- Rabdomyosarkom behandles i henhold til anbefalinger i den avsluttede EpSSG RMS 2005- protokollen. I løpet av 2020 vil det være aktuelt med behandling i henhold til EpSSG-studien FaR-RMS.
- Non-rabdomyosarkom behandles i henhold til EpSSG NRSTS 2005-protokollen.
Lymfeknuter
Målet er å påvise lymfeknuteaffeksjon ved «sampling» og unngå radikalt lymfeknutetoilette. «Sentinel node mapping» med for eksempel methylenblått anbefales.
For spesifikke lokalisasjoner og detaljer forøvrig vises det til Protokoll EpSSG RMS2005, versjon 1.4 international – Dec 2013 (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf) og etter hvert også protokollen FaR-RMS
Kirurgisk behandling av non-RMS bløtvevssarkom (non-RMS STS)
Her er det tilnærmet identiske kirurgiske grunnprinsipper som for RMS. Det vises for øvrig til protokoll EpSSG NRSTS 2005 (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Kirurgisk behandling av bensarkomer
Prinsippet er at tumor må fjernes med fri margin. Det er viktig med nøye planlegging i forkant av inngrepet. Den kirurgiske behandlingen må passe inn i den multimodale behandlingen uten å forsinke øvrig behandling. Se «Nasjonalt handlingsprogram med retningslinjer for utredning, behandling og oppfølging av sarkom» (Helsedirektoratet, 2018).
Strålebehandling
Dersom man ikke oppnår lokal kontroll med kirurgi, kan det være aktuelt med supplerende strålebehandling. Strålebehandling påvirker vekst og utvikling og skal benyttes med varsomhet til barn og ungdom.
Osteogent sarkom er svært lite strålesensitiv, og strålebehandling inngår derfor ikke i rutinebehandlingen, men benyttes kun ved inoperabilitet og i palliativt øyemed.
Ewing sarkom er svært strålesensitiv. Strålebehandling inngår rutinemessig i behandlingen av Ewing sarkom hvis man ikke oppnår fullstendig lokal kontroll med kjemoterapi og kirurgi, det vil si ved marginal og intralesjonell reseksjon, samt ved inoperabilitet. Strålebehandling benyttes dessuten i behandlingen av lunge- og skjelettmetastaser ved Ewing sarkom.
Rabdomyosarkom er strålesensitiv, og strålebehandling inngår som ledd i behandlingen hvis ikke histologien er lite aggressiv og lokalisasjonen gunstig, eventuelt at kirurgien er komplett. Strålebehandling av rabdomyosarkom gjennomføres fortrinnsvis parallelt med postoperativ kjemoterapi.
I gruppen non-rabdomyosarkom er strålefølsomheten variabel, og det er kun enkelte entiteter som synovialt sarkom, «voksen form» for bløtvevsarkom og rabdoid tumor utenfor sentralnervesystemet som behandles med strålebehandling. Både primærtumor og lymfeglandelmetastaser behandles. Strålebehandling gis fortrinnsvis parallelt med postoperativ kjemoterapi (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Oppfølging og etterkontroll
Etter avsluttet behandling for sarkom er det viktig å utføre en sluttevaluering før man starter med regelmessige kontroller. Dette for å evaluere effekten av behandlingen og for å ha et utgangspunkt for senere kontroller. Sluttevalueringen foretas vanligvis 4–6 uker etter avsluttet behandling og omfatter alltid generell klinisk undersøkelse, billeddiagnostikk av affiserte tumorområder, røntgen thorax og blodprøver. Ytterligere detaljer er beskrevet i hver enkelt protokoll. Etterkontrollene skjer vanligvis hver 3. måned de første 2–3 årene, deretter sjeldnere. Hensikten med kontrollene er både å oppdage et eventuelt residiv så tidlig som mulig og å kartlegge eventuelle seneffekter av behandlingen, både på kort og lang sikt. Alle som er behandlet for sarkom, bør følge vanlige kontroller med henblikk på: Høyde og vekt, blodtrykk, Tanner stadium, testikkelvolum, tidspunkt for menarke, skoleprestasjoner, hjertefunksjon, hørsel, nyrefunksjon og eventuelle atferdsproblemer. Detaljer for oppfølging er beskrevet i hver enkelt protokoll.
Se ellers kapittelet om senskader.
Behandling av metastatisk sykdom
Hos enkelte barn er sarkom metastatisk på diagnosetidspunktet. Behandlingen av metastatisk sykdom hos barn baserer seg på de samme prinsipper som ikke-metastatisk sykdom, men kjemoterapien er gjerne enda mer intensiv.
Livsforlengende og palliativ behandling
Ved behov for palliativ behandling hos barn har man ikke lenger et kurativt siktemål, og hensikten med behandlingen vil være å forsøke å lindre plagene så mye som mulig. Dette kan skje ved lavdosekjemoterapi og strålebehandling for å forsinke tumorvekst og lindre smerter, eventuelt smertelindrende medikamenter. Hjemmebehandling og et ordinært liv tilstrebes så lenge som mulig. Viser for øvrig til nasjonale retningslinjer for palliativ behandling hos barn (Helsedirektoratet, 2017).
Nyresvulster
Nefroblastom er den vanligste nyresvulsten i barnealder og kalles til daglig oftest Wilms tumor etter Max Wilms som beskrev den i 1899.
Dette er en ondartet svulst med utgangspunkt i embryonale nyreceller og utgjør ca 90 % av ondartede nyresvulster i barnealder. Opptrer oftest som en enkelt svulst i en nyre, men det kan være multiple svulster, og de kan også opptre i begge nyrer (bilateral svulst). Svulsten diagnostiseres oftest i småbarnsalder og er sjelden hos større barn og ungdommer. Median alder ved diagnose 3,5 år. Over 80 % er under 5 år. Kan også forekomme opp i voksen alder.
Nefroblastom rammer ca 1 av 10 000 barn. Det er ca 6–8 nye tilfeller med nefroblastom i Norge pr år.
Overlevelsen ved nefroblastom er nå gjennomsnittlig ca. 90 % (Pritchard-Jones & Pritchard, 2004).
Genetikk
Nefroblastom forkommer enten sporadisk, i assosiasjon med visse syndromer (se senere) og hos 1–2 % av pasientene som familiær sykdom. Ved familiær sykdom synes genetikken å være kompleks med flere mulige gener involvert.
Kartlegging av genetikk og molekylærbiologi ved nefroblastom er i stadig utvikling. Og man venter at nyere molekylærbiologisk forskning vil kunne identifisere genetiske endringer av betydning for risikoinndeling, behandling og prognose både ved sporadiske og arvelige former. Det vises også til kapittelet Genetikk.
Nefroblastomatose: Betegner en tilstand hvor det i den tidlige nyreutviklingen har blitt værende igjen umodne små foci av embryonale celler (nefrogenic rests) i nyreparenchymet (Beckwith, 1998). Disse vil kunne utvikle seg videre til større hyperplastiske knuter og evt til nefroblastomsvulster. Disse oppdages enten ved at de er så store (hyperplastiske) at de sees som knuter i nyrene på ultralyd ved utredning av nyretumor, eller som tilfeldig funn. De kan også være helt mikroskopiske og finnes ved undersøkelse av gjenværende nyrevev i nyre fjernet grunnet tumor. Det vil da være økt risiko for ny tumor i gjenværende, kontralaterale nyre.
Diagnose
I nesten 50 % av tilfellene har barna ingen symptomer, og svulsten oppdages da tilfeldig enten i forbindelse med andre undersøkelser i helsetjenesten eller ved at foreldre eller andre kjenner at det er en kul i barnets mage. Vel 50 % vil ha magesmerter, få stor, utspilt mage eller ha blod i urinen. Og ca 5 % vil ha tydelig nedsatt allmenntilstand.
Høyt blodtrykk, trombocytose eller hevelse i pungen kan være ledsagende symptomer.
Diagnostikken består av full klinisk undersøkelse hvor man spesielt må være forsiktig ved palpasjon av abdomen, og unngå å trykke hardt på svulsten som kan være skjør. Det er ingen spesifikk tumormarkør knyttet til nefroblastom. Diagnosen stilles ved bildediagnostikk, ultralyd og MR. Fordi 90 % av alle ondartede svulster i nyre er Wilms tumor er det vanlig å starte behandling med cellegift uten forutgående vevsprøve. Vevsprøve forbeholdes de tilfeller hvor diagnosen er usikker etter MR, hvor barnet er yngre enn 7 måneder eller eldre enn 16 år.
Tilstander som disponerer for nefroblastom
Nefroblastom er sterkt assosiert med enkelte syndromer/utviklingsforstyrrelser som:
- Aniridi
- Hemihypertrofi
- Genital/urinveis-midannelser
- Beckwith-Wiedemanns syndrom
- Denys-Drash syndrom
- WAGR syndrom
Pasienter med disse syndromene/utviklingsforstyrrelsene skal kartlegges og tilbys jevnlig kontroll der dette er indisert.
Klassifisering
Klassifiseringen følger den behandlingsprotokollen vi nå bruker:
Umbrella-protokoll SIOP-RTSG 2016. Studien vil åpnes i Norge i løpet av kort tid, men allerede nå brukes den som grunnlag for klassifisering og behandling. Denne protokollen har retningslinjer for behandling av alle typer nyresvulster.
Det er nasjonal enighet om å delta i dette multinasjonale samarbeid for nyresvulster hos barn i regi av SIOP. Det er etablert en organisasjon SIOP Renal Tumour Study Group (www.siop-rtsg.eu) som leder arbeidet. Norge deltar der sammen med de fleste nordiske land. SIOP har gjennomført kliniske studier for nefroblastom fra 1971. Det har vært økende tilslutning av deltagende land. Norge deltok tidligere mest i de engelske nefroblatomstudiene, og sluttet seg, på nasjonal basis, til SIOP-studiene da England også gjorde det fra 2001.
SIOP Renal tumour Study Group har også et nært samarbeid med nyretumorgruppen i COG (Children’s Oncology Group) i USA (http://www.childrensoncologygroup.org/, www.curesearch.org)
Den største forskjell i behandling mellom studiene til SIOP-RTSG og COG er at SIOP-RTSG foretrekker preoperativ kjemoterapi for om mulig å skrumpe tumor før kirurgi og gjøre den lettere håndterbar, mens COG går direkte til primær kirurgi der dette er mulig. De medikamentene som brukes er vinkristin, aktinomycin D og for enkelte grupper også doxorubicin (Kaste et al., 2008; Pritchard-Jones & Pritchard, 2004).
Vår klassifisering bygger på utbredelse etter preoperativ kjemoterapi, og endelig stadiesetting kommer først etter den utsatte kirurgien.
Ved diagnosetidspunktet deles pasientene inn i følgende kategorier:
- lokalisert sykdom
- metastatisk sykdom
- synkron bilateral sykdom
Preoperativ behandling gis ut fra dette. Diagnosen vil oftest baseres på bildediagnostikk alene. Biopsi/cytologi kan bli nødvendig ved usikre funn på MR eller dersom barnet er yngre enn 7 måneder eller eldre enn 16 år, men det er i henhold til den nyeste behandlingsprotokollen altså unødvendig i de fleste tilfeller.
Ultralyd av abdomen er obligatorisk. Metoden kan skille ut cystiske komponeneter og er nyttig for å oppdage små tumores i kontralateral nyre, tumorinnvekst i nyrevene og vena cava, samt metastaser til lever og abdomen. Man får også mål til volumberegning av svulsten ved diagnose. Samme mål gjentas preoperativt etter kjemoterapi.
Rtg.thorax front og side samt CT thorax er også obligatorisk i siste behandlingsprotokoll.
Videre suppleres bildediagnostikken med MR abdomen (alternativt CT abdomen) og evt preoperativ MR eller CT angiografi (for framstilling av blodkar og deres relasjon til tumor).
Nålebiopsi ved nyretumor hos barn skal kun gjøres ved usikkerhet rundt diagnosen etter radiologisk utredning, ved alder yngre enn 7 måneder eller eldre enn 16 år.
Biopsi med Tru-Cut nål utføres av radiolog i generell anestesi, og med patolog til stede for å sikre adekvat vevsmengde. Sykehusets retningslinjer for intervensjon må følges.
Ved bilateral sykdom eller mistanke om nefroblastomatose er det egne anbefalinger med ytterligere utsettelse av kirurgien ved forlenget preoperativ behandling – se protokoll.
Postoperativ behandling: Hele den postoperative behandlingsstratifiseringen hviler på patologens stadieinndeling og histologiske vurdering av operasjonspreparatet etter preoperativ behandling.
| Stadium 1 | Lokalisert til nyren, radikalt fjernet |
| Stadium 2 | Tumorvekst utenfor nyren, men radikalt fjernet |
| Stadium 3 | Tumorvekst utenfor nyren, ikke radikalt fjernet |
| Stadium 4 | Fjernmetastaser (oftest i lungene) |
| Stadium 5 | Tumor i begge nyrer (Bilateral sykdom). |
Histologien deles i lav-, intermediær- og høyrisiko etter bestemte kriterier.
Kombinasjonen av stadium og histologisk risikogruppe bestemmer så hvilken postoperativ kjemoterapi som skal gis, og hvorvidt det også skal suppleres med strålebehandling – se protokoll for detaljer.
Umbrella-protokoll SIOP-RTSG 2016 har som mål gjennom internasjonalt samarbeid å samle pålitelige data som kan brukes til å besvare spørsmål som har direkte klinisk relevans for pasienter. Man ønsker å samle tumorvev samt blod og urin for å lete etter nye markører for å forbedre en risikotilpasset stratifisering av behandlingsintensiteten. Protokollen vil åpnes for å inkludere norske pasienter innenfor kort tid.
Prognosen ved nefroblastom nå er så god at man vil også forsøke å designe behandlingsprogrammer som reduserer risiko for senskader (antracykliner innebærer en risiko for kardiale senskader), og opprettholder de gode behandlingsresultatene.
Andre målsettinger i studien er å forsøke å bedre resultatene ved å optimalisere seleksjonen (riktig stadie/riktig histologi) ved sentral «review» av både histologi og radiologi på alle pasienter
Langtidsoppfølging av barn med nyresvulster
Kirurgi, kjemoterapi og eventuell strålebehandling kan gi senfølger som manifesterer seg kort eller lang tid etter behandling. Nefroblastompasienter har økt risiko for hypertensjon og nedsatt nyrefunksjon. Strålebehandling vil gi senfølger av ulik grad avhengig av omfanget for strålebehandlingen (påvirkning av tarmfunksjon, skoliose, lungeforandringer ved stålebehandling av metastaser, sekundær cancer m.m). Bruk av antracykliner i behandlingen samt evt bestråling av thorax er risikofaktorer for kardiale senskader (Kfr Medisinsk støttebehandling kap. 10) (Wright, Green, & Daw, 2009).
Tilbakefall av nefroblastom
I den nye Umbrella 2016-protokollen har RTSG sammen med COG utarbeidet retningslinjer for behandling av tilbakefall av nefroblastom. Tilbakefallene er sjeldne i en sykdom med så god prognose. De representerer likevel store utfordringer, og økt behandlingsintensitet medfører også risiko for langtidseffekter av behandlingen.
Her brukes som basis de samme medikamentene, vinkristin og actinomycin D (Spreafico et al., 2009) som i primærbehandling med tillegg av andre medikamenter som cyklofosfamid, etoposid og carboplatin. I noen tilfeller anbefales høydosebehandling med stamcellestøtte. I tillegg må mange ha strålebehandling.
Behandling av nefroblastomatosepasienter
Disse pasientene er i økt risiko for å utvikle nefroblastom og skal ha spesielt oppfølgingsprogram og behandling. Det gis ofte langvarig kjemoterapi (minst ett års varighet) inntil forandringene forsvinner eller inntil det blir mulig å fjerne dem med kirurgi. Etter en slik operasjon må det også gis langvarig kjemoterapi.
Andre nyresvulster
I tilegg til en rekke benigne svulster kan det oppstå andre maligne svulster i nyrene; Klarcellet nyresarkom (CCSK), «malign rhabdoid tumour of the kidney» (MRTK), nyrecarcinom samt metastaser fra annen kreftsykdom.
Øvrige retningslinjer som finnes i protokollen
- Behandling for pasienter med primær kirurgi.
- Behandling av bilateralt nefroblasom (gjelder ca 5 % av pasientene).
- Opplegg for behandling av nefroblastomatose.
- Behandling av clear cell sarcoma of the kidney, rhabdoid tumour of the kidney og residiverende sykdom.
- Retningslinjer for oppfølging.
- Behandling av nefroblastom hos voksne.
Kirurgisk behandling av nefroblastom
Det er enighet i det norske barneonkologiske miljøet om nasjonal deltakelse i Umbrella-protokoll SIOP-RTSG 2016, og studien er godkjent av etisk komite. Studien er fortsatt ikke åpnet for inklusjon av norske pasienter, men som «best available treatment» behandler vi i henhold til denne selv om pasientene ikke kan inkuderes i studien. Behandlingen er altså risiko-adaptert og det er viktig å følge protokollen i dens retningslinjer både for kirurgien, stadiesettingen og rapporteringen (sistnevnte kun når pasienter inkluderes). Behandlingsteamet må kjenne behandlingsprotokollen og ha meldeskjemaene for kirurgi for hånden før og/eller under operasjonen.
Tumor i én nyre
Ved operasjon for Wilms’ tumor prioriteres god tilgang fremfor kosmetisk, begrenset snitt. Det anbefales bred, transversal incisjon ovenfor umbilicus og tilgang via laparatomi. Minimal invasiv kirurgi (kikkhull/laparoskopi) er så langt ikke aktuelt hos barn. Bukhulen inspiseres og palperes nøye med tanke på spredning. Moderne billedfremstilling preoperativt gjør det unødvendig å åpne peritoneum/nyrefascien over motsatt sides nyre hvis det ikke er påvist suspekte forandringer i den.
Et generelt prinsipp ved tumorkirurgi er å benytte såkalt «non touch / minimal touch»-teknikk før drenerende kar fra tumor er avstengt. Om mulig bør arterien avklemmes først. Binyren fjernes sammen med nyren hvis ikke tumor er i god avstand fra den. Lymfeknute-sampling er viktig, kfr. behandlingsprotokollen, men radikalt lymfeknutetoilette er (for tiden) ikke indisert. Hvis det foreligger gjennomvekst av kapsel, må en sørge for god margin ut i normalt vev. Heroiske og mutilerende inngrep anbefales imidlertid ikke siden de fleste nyresvulster er både kjemo- og strålesensitive. Preoperativ cytostaticabehandling reduserer sjansene for kapselruptur ved uttak av tumor, men kan vanskeliggjøre bedømmelsen av tumorgrense p.g.a. nekrose/inflammasjon. NB! Sjekk tumors utbredelse på bilder tatt før behandling!
Tumortrombe i vena renalis eller vena cava skal fjernes via venotomi; om tromben når til atriet kan cardiopulmonal bypass være nødvendig.
Nefronsparende tumorreseksjon i stedet for radikal nefrektomi anbefales foreløpig ikke ved ensidig tumor.
Bilateral nyretumor
Ved bilateral tumor bør «nefronsparende» operasjon tilstrebes. Helt avhengig av tumors utbredelse står valget mellom lokal tumorexcisjon bilateralt eller nefrektomi på den ene siden og begrenset reseksjon på den minst affiserte siden. Enukleasjon av små tumores kan være aktuelt, spesielt hvis det er multiple slike i en nyre.
Fjernmetastaser
Lokale levermetastaser skal excideres i forbindelse med primæroperasjonen. Ved utbredte metastaser i leveren skal det tas biopsi. Lungemetastaser skal excideres, men inngrepet kan med fordel utsettes et par uker til resultatene av primæroperasjonen foreligger. Følg for øvrig gjeldende behandlingsprotokoll.
Nevroblastom
Nevroblastom er en ondartet svulst med utgangspunkt i embryonale perifere nerveceller som vanligvis diagnostiseres hos små barn, men sporadiske tilfeller kan også forekomme hos ungdom og unge voksne. I Norge diagnostiseres 6–10 nye tilfeller årlig, og rundt halvparten opptrer som metastatisk sykdom. De fleste nevroblastomer har utgangspunkt i binyrene, men svulsten kan oppstå alle steder der det er sympatisk nervevev (dvs grensestreng og ganglier). Prognosen ved nevroblastom varierer fra nær 100 % overlevelse ved lokal sykdom og gode prognostiske faktorer til rundt 50 % overlevelse hos barn over 1 år med metastatisk sykdom. Dårligst prognose finner vi hos barn > 18 mnd på diagnostidspunktet. Av den grunn er det stor variasjon i behandlingsintensitet hos barn med nevroblastom, fra kun observasjon i de enkleste tilfellene til multimodal intensiv terapi med cellegift, kirurgi, høydosebehandling med stamcellestøtte, stråling og immunterapi hos barn over 1 år med metastatisk sykdom. I Norge behandles nærmest alle barn med nevroblastom etter SIOPEN-protokoller. I SIOPEN samarbeider en rekke land for å oppnå de beste vitenskapsbaserte behandlings- og forskningsprotokoller. Norge er et aktivt medlem av SIOPEN, sammen med 24 andre medlemsland (https://www.siopen.net/).
Diagnose
Symptomer ved nevroblastom er svært varierte og inkluderer skjelettsmerter, fraktur, økt bukomfang, høyt blodtrykk, svette, diaré, nevrologiske utfall, periorbital blødning, Horner syndrom og påvirkning av perifere hematologiske verdier. Diagnosen stilles ved biopsi/cytologi av tumor eller benmargsmetastaser. Forhøyede urinkatekolamin-metabolitter (HVA og VMA), NSE og positive funn ved den nukleærmedisinske undersøkelsen mIBG styrker mistanken om nevroblastom før den cytologiske/histologiske diagnosen foreligger. PET-CT kan gi supplerende informasjon, men anses ikke som nødvendig ved positiv mIBG scan. Det er viktig å sikre nok tumorvev for biologiske analyser (f.eks MYCN-amplifikasjon, kromosomale delesjoner og gains i tumorvev) som kan predikere prognose og nødvendig behandlingsintensitet.
Radiologisk utredning
Siden klassifiseringen baserer seg på den radiologiske utredning med MR og ultralyd (Monclair et al., 2009), er det svært viktig at de preoperative undersøkelsene utføres ved det behandlende sykehus, etter faste protokoller, slik at sammenlikningen av undersøkelsene blir enklere.
Klassifisering
Tidligere stadieinndeling 1–4(S) er med nye protokoller blitt erstattet av den internasjonale INRG-klassifiseringen som forholder seg til risikofaktorer for kirurgi, metastaser og alder (Cohn et al., 2009; Monclair et al., 2009; Pinto et al., 2015);
| L1: | operabel tumor bedømt ut fra radiologiske risikofaktorer |
| L2 | inoperabel tumor bedømt ut fra radiologiske risikofaktorer |
| M: | metastaser til stede |
| MS: | barn under 1 år (18 mnd) med metastaser kun lokalisert til hud, lever og/eller benmarg |
Ung alder og gunstig histologi er forbundet med god prognose, mens biologiske faktorer som segmentale kromosomale aberrasjoner i tumorceller har størst betydning for utfallet av kreftsykdommen. I denne sammenheng er N-Myc amplifikasjon, 11-q delesjon, 1-p delesjon og 17-q gain de viktigste kromosomale avvik. N-Myc amplifikasjon er vanligst hos de minste barna, mens 11-q delesjon har størst betydning for utfallet ved nevroblastom hos eldre barn. Denne erkjennelsen har medført at biologiske faktorer tas hensyn til ved klassifisering og planlegging av behandlingen ved nevroblastomsykdom hos barn.
Behandling
Intensiteten i behandlingen av nevroblastom varierer avhengig av prognostiske faktorer (Pinto et al., 2015). Ytterpunktene demonstreres ved at man ved stadium MS uten livstruende symptomer eller MYCN-amplifikasjon kun observerer, mens man ved stadium M tar i bruk all tilgjengelig målrettet medisinsk behandling (kjemoterapi, kirurgi, høydosebehandling med stamcellestøtte, stråling, vitamin A og immunterapi med spesifikke GD-2 antistoffer). Kjemoterapien består av cisplatin, karboplatin, cyklophosfamid og vinkristin i induksjonsfasen, evt supplert med topotecan, doksorubicin, temozolomid eller irinotecan dersom induksjonsbehandlingen ikke har tilstrekkelig effekt. Nevroblastom har blitt oppfattet som en kreftform der kirurgien er avgjørende. Det er fortsatt tilfelle, men oppdagelsen av biologiske risikofaktorer har medført at enkelte «gunstige» svulster kan observeres uten kirurgi i første omgang. Følgende SIOPEN-protokoller brukes ved de ulike stadier:
| L1: | ingen åpen studie protokoll, kun kirurgisk behandling |
| L2: | protokoll SIOPEN LINES |
| M: | HR-NBL-1 SIOPEN |
| MS: | protokoll SIOPEN LINES / HR-NBL-1 SIOPEN ved MYCN-amplifikasjon |
Internasjonalt er bruken av immunterapi med GD-2 antistoff økende med lovende resultater (Yu et al., 2010). Dinutuximab Beta ble godkjent i Beslutningsforum juni 2019 til behandling av høyrisiko nevroblastom og relapserende/refraktær nevroblastom.
Ved høyrisk nevroblastom kombineres all tilgjengelig effektiv terapi i første behandlingsrunde. Av den grunn er det per i dag lite dokumentert behandling tilgjengelig i residivsituasjoner. Forskningen er intens på dette fagfeltet, og man har håp om at nye behandlingsformer blir dokumentert effektive i nær framtid. Høydose MIBG-behandling med eller uten topotekan-sensitivisering (Gaze et al., 2005) har vist seg effektiv i enkelte situasjoner og kan nå tilbys i Norge i en residiv- eller refraktær situasjon. For høyrisiko nevroblastomer i en residiv/refraktær situasjon der det foreligger informasjon om en ALK abberasjon i tumor, kan det være indisert med behandlingsforsøk med ALK-hemmer (fortrinnsvis en første generasjons ALK hemmer, men noen spesifikke ALK abberasjoner har kjent resistens mot første generasjons ALK hemmere). Denne behandlingen bør fortrinnsvis kombineres med annen tumorrettet behandling. ALK hemmer inngår per 2020 i førstelinjebehandling av høyrisk nevroblastom med ALK abberasjon i USA (Childrens Oncology Group protokoll ANBL 1531). I Europa har man i første omgang ikke inkludert dette i primærbehandlingen, men det brukes i utstrakt grad i en residivsituasjon.
Kirurgi ved nevroblastom
Kirurgisk behandling av nevroblastom vil som regel være radikal fjernelse av tumor. Dette bør skje uten mutilering, skade på vitale organer eller alvorlig annen morbiditet. For svulster med gunstige biologiske markører kan det være akseptabelt å la en liten resttumor bli værende igjen.
Kirurgi er lokalbehandling. Kirurgiens plass i behandlingen må derfor vurderes ut fra om det kan oppnås radikal fjernelse eller ikke. Det er viktig med god kartlegging av tumors utbredelse, både ved diagnosetidspunkt og preoperativt.
Selv om mange nevroblastomsvulster er svært følsomme for kjemoterapi, vil man med unntak av enkelte spedbarn, ikke oppnå at primærtumor forsvinner helt.
Preoperativ kjemoterapi vil ofte være aktuelt, både for å sanere metastaser og for å øke muligheten for radikal fjernelse av tumor. Ved tilstedeværelse av risikofaktorer bør det gis preoperativ kjemoterapi. Ved metastatisk sykdom vil det også gis preoperativ kjemoterapi selv om primærtumor er uten risikofaktorer, med mindre radikal fjernelse av primærtumor anses å være det beste alternativet for vevsdiagnostikk.
Preoperativ kjemoterapi gjør svulstene mindre, mer fibrøse, mindre vaskulariserte og generelt lettere å håndtere. Dette reduserer risiko for spredning peroperativt, og det er akseptabelt å dele tumor under operasjonen og ta den ut bitvis. Det er ofte nødvendig å dele tumor peroperativt fordi tumor ofte omslutter kar som må bevares. Svulstene kan være kraftig adherente til karveggen, men infiltrasjon i selve karveggen er svært sjelden. Etter kjemoterapi er restsvulsten ofte dårlig definert, og det er umulig peroperativt å skille mellom aktivt nevroblastomvev og modent ganglionevrom, fibrose eller arrvev. Derfor vil man ved operasjonen ha som målsetning å fjerne vev i primærtumors utbredelsesområde, men unngå å skade vitale strukturer. Det vanligste er å operere med åpen kirurgi, men for enkelte lokaliserte svulster kan det være aktuelt med laparoskopi eller torakoskopi.
Komplikasjoner relatert til nevroblastomoperasjoner avhenger av utbredelse og lokalisasjon. Ved inngrep på hals og øvre thorax kan det oppstå nerveskader (eks Horners syndrom). I nedre thorax er det en viss risiko for skade på medullas blodforsyning. Retroperitoneale operasjoner kan gi postoperativ diare og ascites. Ved ekstirpasjon av sentrale abdominale nevroblastomer er det risiko for nyreischemi. Nevroblastomkirurgi i bekkenet har risiko for nerveskade, både til blære, tarm og underekstremiteter.
Toksisitet/seneffekter etter nevroblastombehandling
Metastatisk høyrisk nevroblastom krever intensiv behandling, og dermed er risikoen høy for akutt toksisitet og alvorlige seneffekter både forårsaket av grunnsykdommen og behandlingen. Nevroblastomets lokalisering øker ytterligere risikoen for varige seneffekter, siden svulsten ofte infiltrerer nyre eller vokser inn i spinalkanalen. Likevel er det behandlingsinduserte seneffekter av hørsel og nyrefunksjon som er de vanligste følgetilstandene hos langtidsoverlevere etter intensivt behandlet nevroblastom.
Oppfølging/kontroller
Pasientene følges i minst 10 år etter endt behandling. Tumorovervåking baserer seg på ultralyd av tumorlokalisasjon (evt. MR dersom området ikke er tilgjengelig med ultralyd) og urinkatekolaminer. Radiologisk oppfølging av primærtumor kan avsluttes fem år etter endt behandling. Hørsel og nyrefunksjon er av de viktigste seneffektene å monitorere minst 1, 5 og 10 år etter gjennomført behandling.
Leversvulster
2/3 av leversvulstene i barnealder er ondartede. Dette er sjeldne tilstander og utgjør ca 1–2 % av barnekrefttilfellene. Totalt ser vi 0–3 nye tilfeller i Norge pr år.
90 % av de ondartede svulstene i lever er enten hepatoblastom (HB) eller hepatocellulært carcinom (HCC). HB forekommer langt hyppigere enn HCC (Ratio 1,8: 1)
Fordi svulstene er så sjeldne, er det på verdensbasis dannet samarbeidsgrupper som sammen utvikler programmer for diagnostisering og behandling samt forskning på disse svulstene.
I Norge er vi tilsluttet SIOPs levertumorgruppe SIOPEL (https://siope.eu/news/siopel-patientsfamilies-forum-liver-tumours-children/) som har ledet multinasjonale, kliniske studier på leversvulster hos barn siden 1990. Mer enn 30 land deltar i dette SIOPEL–samarbeidet.
Andre store grupper for behandling av leversvulster hos barn er:
- COG (Children’s Oncology Group), USA (www.childrensoncologygroup.org and www.curesearch.org)
- Levertumorgruppen i GPOH (German Society of Pediatric Hematology and Oncology), Tyskland/Østerrike.
- JCCG – The Japanese children’s Cancer group, Japan
Felles for HB og HCC er at de kan produsere og skille ut alfa-fetoprotein (AFP), et glykoprotein som syntetiseres i plommesekk og lever fra tidlig i fosterlivet. Produksjonen stopper normalt ved fødsel, og konsentrasjonen synker fra gjennomsnittlig 50 000 ng/mL ved fødsel til < 20 ng/mL ved 6–8 måneders alder. Variasjonsbredden i AFP-nivå er stor, og det anbefales at man slår opp i tabell i aktuell protokoll. Helt nede på voksne verdier er man først ved 1–2 års alder.
AFP er forhøyet hos over 90 % ved HB og hos vel 50 % ved HCC.
Hepatoblastom
HB er en ondartet svulst med utgangspunkt i embryonale leverceller. Median alder ved diagnose er 1 år. 80 % sees i de 3 første leveår. HB er også beskrevet opp i ungdoms- og voksenalder. HB opptrer oftest som en solitær leversvulst, men kan være multifokal og da affisere store deler av leveren.
Genetikk. HB opptrer oftest sporadisk, og der vet man foreløpig lite om genetiske og molekylærbiologiske faktorers betydning for utvikling, risikoinndelig, behandling og prognose. HB ses også i assosiasjon med visse syndromer hvor noe mer av genetikken er kjent.
Tilstander som disponerer for hepatoblastom
- Isolert hemihypertrofi
- Beckwith-Wiedemann syndrom
- Familiær adenomatøs polypose
Pasienter med disse syndromene/utviklingsforstyrrelsene skal kartlegges og tilbys jevnlig kontroll. Man skal også se nøye etter tegn på disse tilstandene ved diagnostisert HB.
Økt risiko for HB ses også som en assosiasjon til prematuritet med svært lav fødselsvekt.
Diagnose
HB presenterer seg oftest som en asymptomatisk, stor oppfylning i magen hos et ellers friskt barn. Noen har også allmennsymptomer i form av feber, vekttap, anorexi og anemi. Hos noen sees det også koagulasjonsforstyrrelser med blødning ved diagnose.
Laboratorieutredning viser ofte trombocytose og oftest forhøyet AFP-verdi. I sjeldne tilfeller kan også en annen tumormarkør, beta-chorionic gonadotropine (β-HCG) være forhøyet med økt risiko for tidlig pubertetsutvikling.
Utredning
- Grundig anamnese og generell klinisk undersøkelse
- Blodprøver med tumormarkørpanel (AFP, b-HCG, LD, koagulasjonsprøver, evt andre)
- Ultralyd abdomen
- MR eller CT av abdomen
- Rtg thorax og CT thorax
- Evt MR eller CT caput
- Urin til katekolaminbestemmelse (differensialdiagnostikk)
Diagnostisk biopsi
Biopsi gjøres nå som regel som ultralydveiledet tru-cut biopsi som utføres av radiolog i generell anestesi, og med patolog til stede for å sikre adekvat vevsmengde. Det kan også være nødvendig med kirurgisk biopsi hos noen, avhengig av tumors beliggenhet i leveren.
Biopsi er sterkt anbefalt for alle pasienter med mistenkt malign levertumor med mindre det foreligger blødningsforstyrrelser eller kritisk organpåvirkning.
Histologi
Hepatoblastomene deles inn i epiteliale undergrupper: Fetal, embryonal, makrotrabekulær og småcellet udifferensiert (tidligere kalt anaplastisk) og i blandet epitelial og mesenkymal morfologi med eller uten teratoide trekk.
Klassifisering og stadieinndeling
Det har tidligere blitt benyttet flere ulike systemer for klassifisering og stadieinndeling internasjonalt. I USA og flere andre land har det vært vanlig å gjøre primær kirurgi der dette er mulig, og deres inndeling og stadiesetting har vært basert på dette. Dette er forskjellig fra den europeiske tilnærmingen til behandling, noe som har gjort det vanskelig å sammenfatte og sammenligne forskning på dette feltet.
Den pågående studien på leversvulster som Norge også deltar i er imidlertid et samarbeidsprosjekt mellom den europeiske gruppen (SIOPEL), den amerikanske gruppen (COG) og den japanske gruppen (JCCG) kalt PHITT (paediatric hepatic international tumour trial), og man vil i denne studien undersøke kvaliteten på et felles klassifiseringssystem og behandling. Hovedmedikamentet som har vist seg svært effektivt på leverkreft er cisplatin. Dette gis ofte i kombinasjon med doxorubicin og noen ganger også alternerende med carboplatin. Klassifiseringen følger et skjema utviklet av SIOPEL hvor svulstene blir inndelt etter «PRE-Treatment EXTent of disease» (forkortet PRETEXT) Systemet baserer seg på radiologisk påvist affeksjon av leveren ved diagnose i forhold til leverens anatomi med inndeling i 4 seksjoner (8 leversegmenter) samt innvekst i kar og evt tilstedeværelse av metastaser(For beskrivelse av leveranatomi vises det til SIOPEL publikasjoner og protokoller) (Aronson et al., 2005).
Svulstene deles i:
PRETEXT I: kun 1 seksjon involvert
PRETEXT II: 2 «naboseksjoner» involvert
PRETEXT III: 2 seksjoner som ikke ligger inntil hverandre eller 3 «naboseksjoner» involvert
PRETEXT IV: alle 4 seksjoner involvert
Videre inndeles pasientene i very low, low, intermediate og high risk ut fra følgende faktorer:
Tumoregenskaper (ruptur, fokalitet, innvekst i blodkar, vekst utenfor lever, ruptur/blødning)
Metastaser
Barnets alder
AFP-verdi
Tumors resektabilitet
Maligne leversvulster metastaserer først og fremst til lunger og lymfeknuter, men det er også sett spredning til skjelett og CNS. Leversvulstene metastaserer ikke til benmarg.
Behandling
HB er fortsatt en kirurgisk sykdom. Noen få pasienter er primært operable, men strategien for de fleste risikogruppene er preoperativ kjemoterapi med utsatt kirurgi.
Mange pasienter kan få tumor fjernet med leverreseksjon etter preoperativ kjemoterapi. I tillegg kan enkelte, tidligere inoperable pasienter tilbys levertransplantasjon. Kjemoterapien totalt er med på å hindre tilbakefall for de aller fleste. Til sammen har dette gjennom 20 år gitt en betydelig bedret prognose (Perilongo et al., 2009; Perilongo et al., 2004; Pritchard et al., 2000). Se også kapittelet om Kirurgi.
I Norge brukes PHITT-protokollen som klassifiserings- og behandlingsretningslinje, enten barnet deltar i studien eller ikke.
Overlevelse (overall survival) varierer med risikogruppe, og er for standard risiko HB 95 % og for høyrisikogruppen ca 80 % (Zsiros et al., 2013).
Hepatocellulært carcinom
HCC sees oftest hos litt eldre barn (median alder ved diagnose 12 år) og er en langt alvorligere tilstand. HCC forekommer hyppigere i områder hvor hepatitt B virus (HBV) er endemisk, og da oftest hos barn over 5 år. HCC er også assosiert med hepatitt C virus (HCV)
Tilstander som disponerer for HCC
Underliggende leversykdommer som bl.a. tyrosinemi, biliær atresi, idiopatisk neonatal hepatitt, a1‑antitrypsinmangel, nevrofibromatose, ataxia teangiectasia er assosiert med HCC.
Diagnose
Klinisk bilde som ved HB, men positiv AFP kun hos ca 50 %.
Utredning
Som ved HB. I tillegg utvidede tester med fokus på underliggende leversykdom.
Diagnostisk biopsi
Biopsi (ultralydveiledet) bør gjøres hos alle med mindre tumor kvalifiserer for primær kirurgi.
Histologi
Hepatocellulære carcinomer deles histologisk i pediatrisk, adult og fibrolammelær HCC.
Klassifisering og stadieinndeling
Inndeling i PRETEXT-grupper som ved HB – se over.
Maligne leversvulster metastaserer først og fremst til lunger og lymfeknuter, men det er også sett spredning til skjelett og CNS. Leversvulstene metastaserer ikke til benmarg
Behandling
Behandling av HCC er en utfordring, og suksess i behandlingen er totalt avhengig av mulighetene for kirurgi. Dersom tumor er operabel, skal pasienten ha primær kirurgi da HCC er en relativt kjemoresistent tumor. Under halvparten er operable ved diagnose. Pasienter som ikke er operable ved diagnose eller som har mikroskopisk restsykdom etter diagnose vil randomiseres til å motta en av to ulike kjemoterapiregimer. Målet med dette er å finne en behandling som gir reduksjon i tumor slik at flere blir operable, enten ved reseksjon eller ved transplantasjon, og at flere pasienter blir kurert. Tilbakefall av sykdom ved HCC er også en stor utfordring til dags dato.
Generelle kriterier for levertransplantasjon har til nå vært strenge. Hos voksne pasienter aksepteres ikke svulster med diameter > 5 cm eller multifokalitet med mer enn 3 svulster, spesielt ikke hvis en av dem er > 3 cm eller kombinert diameter er > 8 cm (Milan criteria), men man gjør oftest en mer individualisert vurdering for barn. Dog er begrenset sykdom fortsatt et kriterium for suksess også ved transplantasjon. Metastatisk sykdom vil være kontraindikasjon for transplantasjon.
5 års overall survival for HCC i tidligere SIOPEL-protokoller har vært under 30 %. Man har til nå brukt de samme medikamenter ved HCC som ved hepatoblastom, men uten like stor suksess. Man er nå inne i en fase med mer eksperimentell medikamentell behandling i håp om et gjennombrudd for disse pasientene. I PHITT-protokollen sammenlignes behandling med Gemcitabin og Oxaliplatin med konvensjonell HB-behandling. I tillegg er det lagt til Sorafenib i behandlingen for alle pasientene. Dette er et medikament som har vist effektivitet hos voksne pasienter med HCC. I senere studier kan aktuelle medikamenter være bevacizumab, everolimus, sirolimus osv. Lokal intervensjons-behandling direkte mot tumor med radioembolisering eller medikamenter kan også bli aktuelt.
Ved HCC skjer behandlingen etter individuell vurdering og rådføring med internasjonalt fagmiljø.
Overlevelse (overall survival) for HCC er rundt 50 % etter kirurgisk reseksjon/transplantasjon.
Oppfølging
Som for de fleste andre solide svulster følges også barn etter leversvulstbehandling i minst 10 år etter avsluttet behandling og gjerne gjennom puberteten. Barna følges dels for å avdekke residiv, men like viktig er oppfølging med tanke på senskader etter behandlingen.
De to første årene kommer barna til radiologisk undersøkelse hver tredje-fjerde måned. De fleste greier seg med ultralyd abdomen og røntgen thorax, mens noen må ta MR/CT. Man forsøker å unngå CT i kontrolloppfølging grunnet stråledosen. Etter 2 år blir det gradvis lengre intervall mellom kontrolltimene, og vanligvis avsluttes radiologisk oppfølging etter 5 år.
Barn som har fått store kumulative doser kjemoterapi (Cisplatin, Doxorubicin) som kan skade hjerte, nyre eller hørsel får relevante undersøkelser 1, 5 og 10 år etter behandlingsavslutting (kardiotox, hørselstest, GFR).
Kirurgisk behandling av maligne leversvulster
Behandlingen av leverkreft foregår i tett samarbeid mellom barneonkologer, barnekirurger og transplantasjonskirurger. Komplett fjerning av tumor med frie reseksjonsrender er kritisk for langtidsoverlevelse for både hepatoblastom og hepatocellulært carcinom. Leveren har stor regenerasjonsevne, og reseksjoner opp mot 80 % kan tolereres. Hepatoblastomer vil oftest være kjemosensitive, og preoperativ kjemoterapi kan gjøre kirurgien tryggere. Noen svulster kan ved diagnosetidspunktet gi inntrykk av å involvere aller fire segmenter i henhold til Pretext-inndeling. Etter kjemoterapi vil man oftest se at svulsten kan fjernes. Ved multifokale svulster og Pretext IV-svulster vil levertransplantasjon være aktuelt. For enkelte sentralt beliggende svulster som ikke kan fjernes, kan også levertransplantasjon være et alternativ. Komplikasjoner relatert til leverreseksjoner er blødning, gallegangsskader, karskade, luftemboli og leversvikt.
Germinalcellesvulster utenfor CNS
Germinalcellesvulstene (GCT) er en svært heterogen gruppe svulster som varierer betydelig i klinisk presentasjon, lokalisasjon, histologi og biologi. Hos ungdom og unge voksne er de oftest lokalisert i gonadene. I barnealder derimot oppstår de oftest ekstragonadalt i midtlinjen, f eks sacrococcygealt eller i fremre mediastinum. GCT diagnostiseres i alle aldersgrupper fra fosterlivet og opp gjennom oppveksten og står for 3–4 % av krefttilfellene hos barn < 15 år. Modne teratomer kan være underrapportert i slike statistikker (Gobel et al., 1998; Gobel et al., 2000).
Histologi
Svært varierende mikroskopiske bilde. Det ses benigne tumorer, såkalte modne teratomer som kan bestå av mange ulike vevstyper. Det ses også svulster med større eller mindre malignt potensiale, såkalte maligne germinalcellesvulster (MGCT).
Revised Histological Classification of Germ Cell Tumours
- Germinoma
(1) Intratubular
(2) Invasive - Teratoma
a. Mature
b. Immature
c. Malignant teratoma (Teratoma with non germ cell malignant component) - Embryonal carcinoma
- Yolk sac tumour
- Choriocarcinoma
- Gonadoblastoma
- Mixed malignant germ cell tumour
Genetikk
Tilstander som disponerer: Klinefelter syndrom disponerer for mediastinal MGCT.
Ovariale GCT’er viser en signifikant assosiasjon med konstitusjonelle forandringer i kjønnskromosomer som f.eks ved Turner syndrom.
Diagnose
GCT presenterer seg oftest som en asymptomatisk, stor, plasskrevende oppfylning som kan presse på andre organer og gjennom det gi symptomer, for eksempel obstruksjon i luftveiene, vena cava-syndrom, obstipasjon ved bekkensvulster osv. GCT’er skiller ofte ut tumormarkører (AFP og/eller β‑HCG) avhengig av histologisk differensiering. Mønsteret av disse er med på å gi en klinisk diagnose. Husk at AFP normalt er svært høyt i nyfødtperioden og først er nede i «voksne verdier» ved 1–2 års alder. (Mer om AFP, se kapitlet om leversvulster.)
Utredning
- Grundig anamnese og generell klinisk undersøkelse.
- Blodprøver med tumormarkørpanel (AFP, β-HCG og LD. Evt PLAP (placenta-like alkanin fosfatase) som kan være positiv i serum ved germinomer)
- Evt kromosomanalyse (Klinefelter/Turner o.a.)
- Ultralyd abdomen
- MR (CT) av av aktuelle område hvor tumor er lokaliser og av abdomen
- Rtg thorax og CT thorax
- Urin til katekolaminbestemmelse (differensialdiagnostikk)
Klassifisering og stadieinndeling for MGCT varierer mellom ulike studiegrupper og deres protokoller. Protokollen vi følger, UKCCSG GC III, baserer seg på en vanlig TNM-klassifisering, og behandlingsstrategien deler svulstene i lav-, intermediær- og høyrisiko sykdom avhengig av lokalisasjon, utbredelse og nivåer av tumormarkørsekresjon – for detaljer, se protokoll.
De ulike nasjonale og flernasjonale gruppene har ennå ikke klart å samle seg på samme måte som for en del andre solide svulster. Det er derfor flere grupper med litt ulike studier i Europa samt større studier i regi av COG i USA. Det er imidlertid dannet en International Germ Cell Tumour Group som håper å komme i gang med felles studier hvor også randomiserte spørsmål kan bli besvart.
Vi følger nå den engelske behandlingsprotokollen for GCT – UKCCSG GC III (2005) og Children’s Cancer & Leukaemia Group, Germ Cell Group UK- ‘INTERIM GUIDELINES FOR THE TREATMENT OF EXTRACRANIAL GERM CELL TUMOURS IN CHILDREN AND ADOLESCENTS’ fra 2011. Det gjør også Sverige og Danmark, og vi samarbeider i NOPHOs arbeidsgruppe for GCT. I denne protokollen brukes carboplatin i motsetning til cisplatin som de fleste andre studier benytter. Carboplatin gir mindre skade på hørsel og nyrer enn cisplatin gjør, og carboplatin har vist seg i barnealder å være like effektivt som Cisplatin i de to foregående engelske studiene. Carboplatin gis da i kombinasjon med etoposid og bleomycin (J. R. Mann et al., 2000).
Behandling
Modne teratomer fjernes kirurgisk, men skal følges videre med kontroller (klinisk + ultralyd + markører) da de kan residivere både som modent teratom og som MGCT hvis kirurgien har vært inkomplett. Dette gjelder særlig sacrococcygeale svulster hos spedbarn hvor residivraten er på 10–14 %, og hvor residivene ofte er plommesekksvulster.
Immature teratomer. Kirurgi er hovedbehandlingen og er oftest tilstrekkelig alene. Betydningen av økt AFP ved immature teratomer er kontroversiell, og for tiden anbefales at immature teratomer med AFP> 1000 kU/L ved diagnose behandles som MGCT med kjemoterapi i tillegg til kirurgi. De med AFP < 1000 kU/L skal følges tett med ultralyd hver 3. måned og tumormarkørbestemmelse hver 4. til 6. uke. Ved gjenvekst av tumor anbefales ny kirurgi og deretter kjemoterapi.
Alle GCT’er skal ha målt AFP og HCG og gjennomført full utredning. Hvis metastaser ikke er påvist, skal kirurgi vurderes. Inoperable svulster skal, hvis mulig, biopseres. Mutilerende kirurgi skal unngås. Utsatt kirurgi kan vurderes, men skal fortsatt unngås hvis det medfører skader av betydning. GC II-studien har vist at bare 2 av 45 barn hadde viabelt tumorvev ved utsatt kirurgi etter gjennomført kjemoterapi. De øvrige hadde nekrose/fibrose eller modent/umodent teratom. Slike pasienter bør heller følges tett for å se om utviklingen nødvendiggjør ytterligere behandling.
Lavrisiko MGCT defineres som alle stadium 1 svulster (kfr protokoll) Disse får ingen postoperativ kjemoterapi, men følges tett med ukentlige kontroller til normalisering av tumormarkører og deretter kontroll hver måned.
Intermediær risiko MGCT er alle seminomer/germinomer uansett AFP-verdi, de fleste stadium 2 og 3 svulster med AFP < 10 000 kU/L unntatt thoracale svulster hvor st. 2, 3 og 4 alle er høyrisiko, mens testis st 4 < 5 år er intermediær.
Disse får 4 kurer med kjemoterapi.
Høyrisiko MGCT er alle med AFP > 10 000 kU/L unntatt stadium 1 svulster og untatt testissvulster hos gytter < 5 år. Alle thoracale svulster st 2,3 og 4 inngår i gruppen uavhengig av AFP nivå.
Disse får 6 kurer med kjemoterapi.
Strålebehandling er ikke en integrert del av behandlingsopplegget for ekstrakraniell GCT i barnealder. Men strålebehandling kan være aktuell ved tilbakefall. Dysgerminom i ovariet og seminom i testis er svært strålefølsomme, og strålebehandling kan være aktuelt i utvalgte situasjoner. Men dette vil være etter individuell vurdering hos erfaren stråleonkolog.
Residivbehandling
Protokollen gir også anbefalinger for behandling ved residiv. Kjemoterapien er da vesentlig cisplatinbasert.
Prognose
Prognosen er generelt god. Gjennom de siste 20 år har prognosen for maligne GCT’er bedret seg betydelig. Mye av dette kan tilskrives bedret multimodal tilrettelegging av behandlingen samt ikke minst, introduksjonen av cisplatin i kjemoterapien. Der hvor man kan fjerne tumor radikalt, er overlevelsen nå > 80 %, men enkelte undergrupper som choriocarcinom kan ha dårligere prognose.
Oppfølging
De fleste barn følges i minst 10 år etter avsluttet behandling og gjerne gjennom puberteten. Barna følges dels for å avdekke residiv da disse ofte kan behandles med godt resultat. Like viktig er også oppfølging med tanke på senskader etter behandlingen.
Den første tiden følges de med forhøyede tumormarkører tett for å se at disse faller til normale verdier. Deretter månedlige tumormarkørkontroll i et år, så hver annen måned det andre året og hver 3. måned det tredje året.
De to første årene kommer barna også til radiologisk undersøkelse hver tredje måned. De fleste greier seg med ultralyd abdomen og røntgen thorax, mens noen må ta MR/CT. Man forsøker å unngå CT i kontrolloppfølging grunnet stråledosen. Etter 2 år blir det gradvis lengre intervall mellom kontrolltimene, og vanligvis avsluttes radiologisk oppfølging etter 5 år.
Barn som har fått store kumulative doser kjemoterapi som kan skade hjerte, lunger, nyre eller hørsel får relevante undersøkelser 1, 5 og 10 år etter behandlingsavslutting (kardiotox, lungefunksjonstest, hørselstest, GFR). Man er også oppmerksom på stråleinduserte skader og samarbeider med ortoped, pedagog og andre dersom tegn til stråleskader oppdages. Man er også oppmerksom på endokrinologiske bivirkninger og samarbeider tett med barneendokrinolog, eventuelt gynekolog.
Retinoblastom (RB)
Epidemiologi
Retinoblastom er en sjelden svulst som oppstår i ett eller begge øyne fra umodne retinaceller hos barn. Insidens av RB i Norge er omtrent 1/15 000 levende fødte barn (Gregersen et al., 2016). Det oppdages ca. 4 nye tilfeller per år og det er like mange gutter som jenter som får RB. Selv om RB kan inntreffe i alle aldre, oppstår det oftest før 2 års alderen, og ca. 95 % av tilfellene er diagnostisert før 5 års alder. Opp mot 98 % av alle barn med RB blir helbredet. Dette er et resultat av tidlig diagnostikk og gode behandlingsmuligheter.
Klassifikasjon
Nå klassiffiseres RB hovedsakelig i henhold til den Internasjonale klassifiseringen; ABCDE (Murphree, 2005). Tidligere benyttet man også Reese-Ellsworth (Yousef et al., 2015), og TNM (Rosengren, Monge, & Flage, 1989). Det kan være en fordel å benytte alle tre klassifikasjoner slik at det kan gjøres sammenlikninger med tidligere publiserte materialer.
Veksttyper
Endofytisk: Vokser fra netthinneoverflaten og inn mot eller inn i glasslegemet. Tumorens overflate er oppblåst og dårlig avgrenset med atypiske kar (figur 1).
Eksofytisk: Vokser utover og løfter opp netthinnen til evt. amotio (figur 2).
Diffust infiltrerbar: Dette er en uvanlig form som ofte diagnostiseres sent på grunn av differensialdiagnostiske vanskeligheter. De aller fleste pasientene er eldre enn ved de to andre formene. Tumor er vanligvis ensidig og oppstår spontant (Kim, Abramson, & Dunkel, 2007).
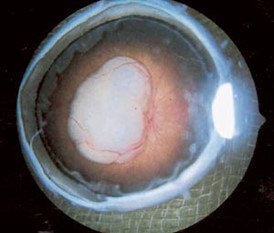
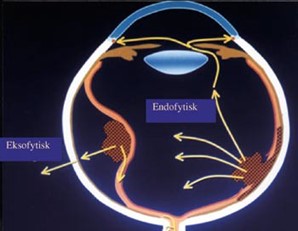
Patogenese, genetikk og etiologi
Retinoblastom skylles mutasjon av begge alleler av RB1 genet, som er lokalisert på kromosom 13 bånd q14 og er et stort gen som omfatter 200 000 basepar (Friend et al., 1986; Kohno, Kitajima, Sasaki, & Takahashi, 2016). RB1 er et tumor-suppressorgen. Pasienter med den arvelige formen har en medfødt germline mutasjon på et av allelene (first hit), som enten kan være nyoppstått eller nedarvet fra foreldrene (10 %). Penetransen ved RB1 mutasjoner er høy, men det finnes tilfeller med nedsatt penetrans (samlet penetrans er ca. 95 %). Tumor oppstår som en somatisk mutasjon i det andre allelet (second hit) (Cook et al., 2017). De fleste av de arvelige tilfellene er multifokale og bilaterale, og gjennomsnittsalderen ved diagnose er 15 mnd. Ved de ikke arvelige, sporadiske tilfellene er tumor oppstått som følge av to somatiske mutasjoner. Disse er oftest unilaterale og unifokale, og gjennomsnittsalderen ved diagnose er ca. 24 mnd.
Ved den arvelige varianten kan det også forekomme tumor i corpus pineale. Dette kalles trilateralt retinoblastom, og MR cerebrum us. er viktig for å utelukke dette.
Symptomer og tegn
Ved undersøkelse av barn, både ved nyfødtscreeningen og på helsestasjonene, bør den minste mistanke om leukokori eller skjeling medføre umiddelbar henvisning til øyelege (Dimaras et al., 2012; Sandven-Thrane, 2012). De vanligste debutsymptomene er:
- Leukokori (hvit refleks): 56–69 %
- Strabisme (skjeling): 20–35 %
- Nedsatt syn
- Glaukom
- Rødt øye
- Uveitter
- Unilateral mydriasis
Utredning
Øyeundersøkelse i våken tilstand: Sjekk av visus, synsfelt, skjeling, pupillerefleks og øyebevegelse.
Øyeundersøkelse i full narkose: Undersøkelse av øyebunnen i maksimal mydriasis og med oraimpresjon. Bruk av ultralyd for måling av tumorstørrelse, vurdere innhold av kalk og ev. gjennomvekst av øyeveggen. Klinisk foto av øyebunnen. Biopsi er ikke anbefalt p.g.a. risiko for sekundær spredning.
Blodprøver: Hb, leukocytter med differesialtelling, trombocytter, natrium, kalium, kreatinin, ASAT, ALAT, LD, CRP, leverfunksjonsprøver, serologiske undersøkelser for CVM, EBV og Varicella og prøver til genetikk.
Full somatisk undersøkelse.
MR orbita og cerebrum: Framstiller godt tumor fra vevet omkring. Ser etter mulig ekstraokulær vekst og corpus pinealesvulster. Tumorvevet fremstilles som moderat hyperintensivt på T1 og hypointensivt på T2 bilder. MR kan også skille mellom tumorvev, blødning og eksudat.
Barnet og familien henvises til genetisk samtale/veiledning.
Ev. CT orbita: Fremstiller kalk i tumor, god på påvisning av vekst utenfor øyet.
Ved ev. enukleasjon: Ta ferskt tumorvev til mutasjonsanalyse før formalinfiksering.
Behandling
Innledning
Både diagnostikk og behandling av retinoblastom er multimodal og omfattende og skal gis ved sykehus med spesialkompetanse i slik behandling.
Øyeavdelingen og Barneavdeling for kreft og blodsykdommer ved Oslo Universitetssykehus har landsfunksjon for retinoblastom. Alle retinoblastomene utredes og behandles primært her. Alle oftalmologiske kontroller under behandlingen gjøres her.
Små svulster kan behandles med lokalbehandling, som kryobehandling, laser behandling eller stråleplate, utført ved øyelege. Ved større svulster kan det være aktuelt med kjemoterapi, og i noen tilfeller må man fjerne øyet (enukleasjon). Ekstern stråling brukes sjelden i dag.
Om det skal gis konvenskonell, intravenøs kjemoterapi, kan dette gis på lokalsykehus med kompetanse på kjemoterapi. Mellom kurene kan barnet gjerne være hjemme, men må da ha tett kontakt med lokalsykehuset for blodprøvekontroller. Støttebehandling i form av transfusjoner og eventuell antibiotikabehandling, gis på lokalsykehuset etter vanlige retningslinjer.
Kjemoterapi
Ved behov for kjemoterapi, er primærbehandlingen nå ofte cytostatika (Melfalan) direkte inn i a. ophthalmica (IAM). Dette har vist goderesultater hos de fleste (Radros et al., 2018). I tillegg unngår man bivirkningene som ved systemisk behandling. Denne behandlingen brukes som volumreduserende tiltak før annen behandling (Shields et al., 2004; Shields, Meadows, Shields, Carvalho, & Smith, 2001). Nødvendigheten av tilleggsbehandling vurderes ved øyeundersøkelse i narkose i tilslutning til hver 2. kur. Om ikke IAM behandling er aktuelt eller er kontraindisert (for eksempel hos de aller minste barna), kan det gis systemisk kjemoterpi med Vincristin, Carboplatin og Etoposid med 3 ukers mellomrom. Det gis vanligvis 3–6 kurer.
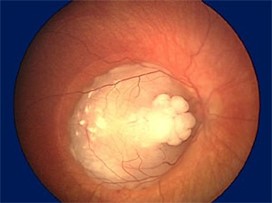
Kryobehandling
Vevet fryses til –70 °C. Benyttes ved mindre perifere svulster (mindre enn 6 mm i diameter og 3 mm høyde).
Laserbehandling
Det finnes ulike typer laser som benyttes for å ødelegge blodforsyningen til tumor. Benyttes ved små svulster (mindre enn 4–7 mm i diameter og 3 mm høyde) særlig de som ligger langt bak i øyet. Kan også gis i tilslutning til kjemoterapi primært, eller ved små residiv.
Strålebehandling
Retinoblastom er en strålefølsom tumor (Kim et al., 2007; Swanson et al., 2011)
Lokal strålebehandling med plate (Ru-106 (ruthenium) eller I-125 (jod)). Disse avgir betastråler respektive lavenergetiske gammastråler.
Indikasjoner:
Tumor < 15 mm i diameter og 3–7 mm tykk.
Tumores som er minst 2 mm fra n. opticus eller macula.
Tumores med lokal infiltratrasjon i glassvæsken.
Turmores som ikke har svart på annen behandling eller ved residiv.
Dosen er 35–40 Gy mot tumors apeks.
Ekstern strålebehandling
Indikasjon
Ved residiv hvor man vil bevare et seende siste øye, hvor annen behandling ikke har virket, eller ved spredning ut i glasslegemet.
Dosen er 35–40 Gy, 1,5–2 Gy 5 dager per uke i 5 uker.
Om det er aktuelt med strålebehandling, gis dette i form av protonbestråling som har kortere strålerekkevidde og derfor ikke skader tilstøtende vev like mye som fotoner.
Enukleasjon
Benyttes enten når svulsten ved diagnosen er så stor at det ikke er mulig å bevare visus, ved residiv man ikke får under kontroll, eller ved tegn til metastaser. Ved enukleasjon er det viktig at man får med seg så mye av n.opticus som mulig. Dersom det ved histologisk undersøkelse av det fjernede øye finnes tegn til gjennomvekst i n. opticus, eller ved tegn til infiltrasjon gjennom sklera, er det nødvendig med kjemoterapi eller strålebehandling (Nordiske retningslinjer for retinoblastom, 2016).
Prognose
Overlevelsen av RB er 98 %. Lokalt residiv er vanlig, men kommer svært sjelden etter at barnet har fylt 3 år.
Prognostiske faktorer mhp residiv:
Tumors størrelse og utbredelse.
Eventuelt forhøyet intraokulært trykk ved diagnosen.
Alder ved diagnose. Prognosen med henblikk på residiv er bedre ved diagnose mellom 2 og 7 år enn for pasienter som diagnostiseres tidligere eller senere.
Risiko for nye retinale tumores er (Abramson, Greenfield, & Ellsworth, 1992)
58 % hvis diagnosen stilles < 3 mnd alder
45 % hvis diagnosen stilles mellom 3 og 6 mnd alder
14 % hvis diagnosen stilles > 6 mnd alder
De som har arvelig retinoblastom, har større risiko for å utvikle sekundære maligniteter, dvs. sarkom i ben og bløtvev og kutane melanomer (Kleinerman et al., 2005; Kleinerman et al., 2012; Turaka, Shields, Meadows, & Leahey, 2012; Wong et al., 2014). 70 % av tilfellene oppstår i hodet (innen strålefeltet) ved ekstern strålebehandling Dette er avhengig av dose/respons og alder. Disse tumortypene andre steder i kroppen er ikke relatert til strålebehandling, men til mutasjonene i RB1-genet.
Utvikling av ev. komplikasjoner i øyet
Fra gjenværende tumor og arrvev
Vaskulære anomalier
Blødninger
Forandringer på retina
Glaukomutvikling
Ofte utvikling av pussdannelse ved enukleasjon.
Forandringer etter strålebehandling
Kosmetiske
Sekundær atrofi av orbita
Atrofi av fett og bindevev – øyet blir innsunket
Stråleretinopati og opticusnevropati
Utvikling av katarakt
Ofte store plager med tørre øyne, trenger kunstig tårevæske
Videre kontroller
Barn som er behandlet for RB bør gå til hyppige kontroller både hos pediater og oftalmolog med henblikk på:
Residiv
Ny cancer, primær eller sekundær (i strålefeltet)
De bør også følges opp på lokal BUP ved behov.
Øyelegekontroll:
Barn som har gjennomført behandling for retinoblastom bør kontrolleres:
Hver 2. mnd et år etter siste behandling. Deretter individuell nedtrapping. Avhengig av antatt arvelighet og gjenværende tumorforandringer i øyet/øynene skal noen ha kontroll hos øyelege hele livet.
Barn til foreldre med bilateralt retinoblastom bør kontrolleres:
Så raskt som mulig etter fødsel (innen maks 2–3uker)
Hver 2.–3. mnd til 2 års alder.
Hver 4. mnd til 4 års alder
Hver 6. mnd til 7–8 års alder
Søsken til barn med retinoblastom og barn til foreldre med unilateralt retinoblastom bør kontrolleres:
Hver 2.–3. mnd første året.
Hver 4. mnd til 4 års alder.
Hver 6. mnd til 7–8 års alder